Beckwith-Wiedemann syndrome (BWS) is a growth disorder that can affect several parts of the body. Babies and children are larger than normal usually until age 8, when growth slows down, resulting in an average height in adults. Symptoms may include one side or area of the body growing more than the other side (asymmetric growth or hemihyperplasia ), omphalocele or other abdominal wall defect at birth, low blood sugar (hypoglycemia) in infancy, an abnormally large tongue (macroglossia), abnormally large abdominal organs, creases or pits in the skin near the ears, and kidney abnormalities. Affected children have an increased risk to develop tumors, particularly a rare form of kidney cancer called Wilms tumor , a cancer of muscle tissue called rhabdomyosarcoma, and a form of liver cancer called hepatoblastoma . Some people only have one symptom while others may have many of the symptoms.
Beckwith-Wiedemann syndrome (BWS) is a genetic disorder characterized by overgrowth, tumor predisposition and congenital malformations. Clinical description Patients tend to grow at an increased rate during the 2nd half of pregnancy and in the first few years of life; adult heights are typically in the normal range. Abnormal growth may also manifest as hemihyperplasia and/or macroglossia (leading to difficulties in feeding, speech and infrequently, sleep apnea). Hypoglycemia is reported in 30-50% of neonates. A recognizable facial gestalt is common and often normalizes by adulthood. In addition to macrosomia, macroglossia, hemihyperplasia and hypoglycemia, characteristic findings may include omphalocele/umbilical hernia/diastasis recti, embryonal tumor, anterior earlobe crease(s) and posterior helical pit(s), nevus flammeus or other vascular malformations, visceromegaly involving abdominal organs, fetal adrenocortical cytomegaly (pathognomonic), renal abnormalities, positive family history, and rarely cleft palate.
Summary Clinical characteristics. Beckwith-Wiedemann syndrome (BWS) is a growth disorder variably characterized by neonatal hypoglycemia, macrosomia, macroglossia, hemihyperplasia, omphalocele, embryonal tumors (e.g., Wilms tumor, hepatoblastoma, neuroblastoma, and rhabdomyosarcoma), visceromegaly, adrenocortical cytomegaly, renal abnormalities (e.g., medullary dysplasia, nephrocalcinosis, medullary sponge kidney, and nephromegaly), and ear creases/pits. BWS is considered a clinical spectrum, in which affected individuals may have many of these features or may have only one or two clinical features. Early death may occur from complications of prematurity, hypoglycemia, cardiomyopathy, macroglossia, or tumors. However, the previously reported mortality of 20% is likely an overestimate given better recognition of the disorder along with enhanced treatment options. Macroglossia and macrosomia are generally present at birth but may have postnatal onset.
Beckwith-Wiedemann syndrome is a condition that affects many parts of the body. It is classified as an overgrowth syndrome, which means that affected infants are considerably larger than normal (macrosomia) and tend to be taller than their peers during childhood. Growth begins to slow by about age 8, and adults with this condition are not unusually tall. In some children with Beckwith-Wiedemann syndrome, specific parts of the body on one side or the other may grow abnormally large, leading to an asymmetric or uneven appearance. This unusual growth pattern, which is known as hemihyperplasia, usually becomes less apparent over time.
A number sign (#) is used with this entry because Beckwith-Wiedemann syndrome (BWS) can be caused by mutation or deletion of imprinted genes within the chromosome 11p15.5 region. Specific genes involved include p57(KIP2) (CDKN1C; 600856), H19 (103280), and LIT1 (KCNQ1OT1; 604115). Hypermethylation and variation in the H19/IGF2-imprinting control region (ICR1; 616186) on chromosome 11p15.5, which regulates imprinted expression of H19 and IGF2 (147470), is also associated with BWS. See also Silver-Russell syndrome (SRS; 180860), which is caused by hypomethylation defects at 11p15. Description Beckwith-Wiedemann syndrome is a pediatric overgrowth disorder involving a predisposition to tumor development.
PMID 24402832 . ^ Tamiya T, Nakashima H, Ono Y, et al. (2000). "Spinal atypical teratoid/rhabdoid tumor in an infant". ... (February 2003), "A unique occurrence of a cerebral atypical teratoid/rhabdoid tumor in an infant and a spinal canal primitive neuroectodermal tumor in her father", Journal of Neuro-Oncology , 61 (3): 219–225, doi : 10.1023/A:1022532727436 , PMID 12675315 , S2CID 2766929 ^ Ho DM, Hsu CY, Wong TT, Ting LT, Chiang H (2000). "Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma".
ACTH-secreting pituitary tumors associated with Cushing syndrome are surgically removed; nonsecreting pituitary adenomas are treated by transsphenoidal surgery. Proton pump inhibitors or H 2 -receptor blockers reduce gastric acid output caused by gastrinomas. ... Well-Differentiated Tumors of the Gastro-Entero-Pancreatic (GEP) Tract Gastrinoma Medications that can control some of the GEP hormone excess-dependent features of MEN1 syndrome and thus prevent severe and sometimes life-threatening morbidity in MEN1 syndrome include proton pump inhibitors or H 2 -receptor blockers to reduce gastric acid output [Jensen 1999].
A number sign (#) is used with this entry because multiple endocrine neoplasia type I (MEN1) is caused by heterozygous mutation in the MEN1 gene (613733) on chromosome 11q13. Description Multiple endocrine neoplasia type I (MEN1) is an autosomal dominant disorder characterized by varying combinations of tumors of parathyroids, pancreatic islets, duodenal endocrine cells, and the anterior pituitary, with 94% penetrance by age 50. Less commonly associated tumors include foregut carcinoids, lipomas, angiofibromas, thyroid adenomas, adrenocortical adenomas, angiomyolipomas, and spinal cord ependymomas. Except for gastrinomas, most of the tumors are nonmetastasizing, but many can create striking clinical effects because of the secretion of endocrine substances such as gastrin, insulin, parathyroid hormone, prolactin, growth hormone, glucagon, or adrenocorticotropic hormone (summary by Chandrasekharappa et al., 1997). Familial isolated hyperparathyroidism (see 145000) occasionally results from the incomplete expression of MEN1 (summary by Simonds et al., 2004).
A rare inherited cancer syndrome, characterized by the development of multiple neuroendocrine tumors of the parathyroids, gastro-entero-pancreatic tract, and anterior pituitary gland, and less commonly the adrenal cortical gland, thymus and bronchi, with other non-endocrine tumors in some patients. Epidemiology Prevalence of multiple endocrine neoplasia type 1 (MEN1) is estimated to range between 1/10,000-30,000. The sex ratio is equal. Clinical description Tumors can develop at any age with 95% of patients developing clinical symptoms by the 5th decade. Parathyroid tumors are the most common (95% of patients), followed by pancreatic islet tumors (40%) and anterior pituitary tumors (30%). Pancreatic neuroendocrine tumors include gastrinoma (50%), insulinoma (33%), glucagonoma (5%), vasoactive intestinal peptide (VIP)-oma, pancreatic polypeptide (PP)-oma and non-functioning tumors, and are associated with high levels of morbidity and mortality.
Multiple endocrine neoplasia is a group of disorders that affect the body's network of hormone-producing glands called the endocrine system. Hormones are chemical messengers that travel through the bloodstream and regulate the function of cells and tissues throughout the body. Multiple endocrine neoplasia typically involves tumors (neoplasia) in at least two endocrine glands; tumors can also develop in other organs and tissues. These growths can be noncancerous (benign) or cancerous (malignant). If the tumors become cancerous, the condition can be life-threatening. The major forms of multiple endocrine neoplasia are called type 1, type 2, and type 4.
Multiple endocrine neoplasia, type 1 (MEN1) causes the growth of tumors in both the endocrine system (the body's network of hormone-producing glands) and non-endocrine system. Symptoms of MEN1 include tumors of the parathyroid gland , the pituitary gland , and the pancreas, although other glands may be involved as well. These tumors are often "functional" and secrete excess hormones, which can cause a variety of health problems. Additional signs and symptoms of MEN1 may depend on the type of tumor present and which hormones are being secreted. The most common symptoms are cause by an overactive parathyroid gland and may include kidney stones; thinning of bones; nausea and vomiting; high blood pressure (hypertension); weakness; and fatigue.
Facioscapulohumeral muscular dystrophy Other names Landouzy–Dejerine muscular dystrophy, FSHMD, FSH A diagram showing in red the muscles commonly involved in FSHD Pronunciation / f æ ʃ iː oʊ s k æ p j ə l oʊ ˈ h j u m ər əl / Specialty Neurology , neuromuscular medicine Symptoms Facial weakness, scapular winging, foot drop Usual onset Adolescence Duration Long term Types FSHD1, FSHD2 Causes Genetic (inherited or new mutation) Diagnostic method Genetic testing Differential diagnosis Limb-girdle muscular dystrophy (especially calpainopathy ), Pompe disease , mitochondrial myopathy , polymyositis [1] Management Physical therapy, bracing, reconstructive surgery Frequency 1 in 8333 to 1 in 15000 [1] Facioscapulohumeral muscular dystrophy ( FSHD ) is a type of muscular dystrophy that preferentially weakens the skeletal muscles of the face (Latin: facio ), those that position the scapula ( scapulo ), and those in the upper arm , overlying the humerus bone ( humeral ). [1] Weakness of the scapular muscles causes an abnormally positioned scapula ( winged scapula ). ... Muscles can be scored based on the degree of fat infiltration. [1] References [ edit ] ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al am Wagner, Kathryn R. ... PMC 2965761 . PMID 21060811 . ^ a b c d e f g h i j k l m Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, Miller DG, Tapscott SJ, Tawil R, Frants RR, van der Maarel SM (19 August 2010). ... PMC 2265642 . PMID 17924332 . ^ a b c d e f g h Mul, Karlien; Lassche, Saskia; Voermans, Nicol C; Padberg, George W; Horlings, Corinne GC; van Engelen, Baziel GM (June 2016).
A number sign (#) is used with this entry because facioscapulohumeral muscular dystrophy-2 (FSHD2) shows digenic inheritance. It is caused by the combination of a heterozygous mutation in the SMCHD1 gene (614982) on chromosome 18p and presence of a haplotype on chromosome 4 that is permissive for DUX4 (606009) expression. Description Facioscapulohumeral muscular dystrophy is a form of muscular dystrophy characterized by muscle weakness that first affects the facial muscles and upper extremities, later progressing to involve the lower extremities. The pattern of weakness is usually asymmetric (summary by Lemmers et al., 2012). FSHD1 (158900), which is clinically indistinguishable from FSHD2, is associated with contraction of the D4Z4 macrosatellite repeat (see 606009) in the subtelomeric region of chromosome 4q35.
Facioscapulohumeral muscular dystrophy is a disorder characterized by muscle weakness and wasting (atrophy). This condition gets its name from the muscles that are affected most often: those of the face (facio-), around the shoulder blades (scapulo-), and in the upper arms (humeral). The signs and symptoms of facioscapulohumeral muscular dystrophy usually appear in adolescence. However, the onset and severity of the condition varies widely. Milder cases may not become noticeable until later in life, whereas rare severe cases become apparent in infancy or early childhood. Weakness involving the facial muscles or shoulders is usually the first symptom of this condition.
A rare neuromuscular disease characterized by progressive muscle weakness with focal involvement of the facial, shoulder and limb muscles. Epidemiology Facioscapulohumeral muscular dystrophy (FSHD) is a rare familial disease with an estimated prevalence from 1/8,000 to 1/20,000. It is the third most common form of hereditary myopathy. Clinical description Onset occurs at any age. Disease progression is usually slow but some patients display periods of stability followed by periods of rapid deterioration. Early onset of FSHD is associated with more widespread muscle weakness.
A number sign (#) is used with this entry because facioscapulohumeral muscular dystrophy-1 (FSHD1) is associated with contraction of the D4Z4 macrosatellite repeat (see 606009) in the subtelomeric region of chromosome 4q35. In unaffected individuals, the D4Z4 array consists of 11 to 150 repeat units (corresponding to EcoRI fragments of 41 to 350 kb), whereas FSHD patients have contraction of the repeat units from 1 to 10 (corresponding to EcoRI fragments of 10 to 35 kb). Borderline fragment sizes (35 to 40 kb) must be interpreted with caution (summary by Mostacciuolo et al., 2009). The genetics of FSHD is, however, complex, and detection of a D4Z4-reduced allele may not be sufficient for diagnosis (see DIAGNOSIS below and Scionti et al., 2012). Description Facioscapulohumeral muscular dystrophy is the third most common hereditary disease of muscle after Duchenne (DMD; 310200) and myotonic (160900) dystrophy.
Facioscapulohumeral muscular dystrophy is a disorder characterized by muscle weakness and wasting (atrophy). This condition gets its name from the areas of the body that are affected most often: muscles in the face (facio-), around the shoulder blades (scapulo-), and in the upper arms (humeral). The signs and symptoms of facioscapulohumeral muscular dystrophy usually appear in adolescence. However, the onset and severity of the condition varies widely. Facioscapulohumeral muscular dystrophy results from a deletion of genetic material from a region of DNA known as D4Z4. This region is located near one end of chromosome 4 . It is inherited in an autosomal dominant pattern.
Summary Clinical characteristics. Facioscapulohumeral muscular dystrophy (FSHD) typically presents with weakness of the facial muscles, the stabilizers of the scapula, or the dorsiflexors of the foot. Severity is highly variable. Weakness is slowly progressive and approximately 20% of affected individuals eventually require a wheelchair. Life expectancy is not shortened. Diagnosis/testing. The diagnosis of FSHD1 is established in a proband with characteristic clinical features by identification of a heterozygous pathogenic contraction of the D4Z4 repeat array in the subtelomeric region of chromosome 4q35 on a chromosome 4 permissive haplotype. The diagnosis of FSHD2 is established in a proband by identification of hypomethylation of the D4Z4 repeat array in the subtelomeric region of chromosome 4q35 on a chromosome 4 permissive haplotype. Hypomethylation of the D4Z4 repeat array can be due to a heterozygous pathogenic variant in SMCHD1 or DNMT3B .
Jardine et al. (1994) described 7 affected individuals, 3 men and 4 women, in a 2-generation family segregating a scapulohumeral muscular dystrophy. Weakness began in the shoulders between 12 and 40 years of age. There was no distal weakness in the upper or lower extremities and there were no sensory abnormalities. In several cases, there was marked asymmetry with weakness on the right side more than on the left. There was no demonstrable facial weakness in any of the affected individuals. Male-to-male transmission was not observed. There was only minimal elevation of creatine kinase in some individuals.
PMID 21674591 . ^ Sano K, Satoh K, Atarashi R, Takashima H, Iwasaki Y, Yoshida M, Sanjo N, Murai H, Mizusawa H, Schmitz M, Zerr I, Kim YS, Nishida N (2013-01-25). ... PMID 8638007 . ^ Ukisu R, Kushihashi T, Kitanosono T, Fujisawa H, Takenaka H, Ohgiya Y, Gokan T, Munechika H (February 2005). ... PMID 15671380 . ^ Young GS, Geschwind MD, Fischbein NJ, Martindale JL, Henry RG, Liu S, Lu Y, Wong S, Liu H, Miller BL, Dillon WP (June–July 2005). ... PMID 25667875 . ^ Mizutani T, Okumura A, Oda M, Shiraki H (February 1981). "Panencephalopathic type of Creutzfeldt-Jakob disease: primary involvement of the cerebral white matter" . ... PMID 9204296 . ^ Ae R, Hamaguchi T, Nakamura Y, Yamada M, Tsukamoto T, Mizusawa H, Belay ED, Schonberger LB (March 2018).
A number sign (#) is used with this entry because familial Creutzfeldt-Jakob disease can be caused by mutation in the prion protein gene (PRNP; 176640). Gerstmann-Straussler disease (GSD; 137440) and familial fatal insomnia (FFI; 600072) are 2 other allelic inherited prion diseases caused by mutation in the PRNP gene. Description The human prion diseases occur in inherited, acquired, and sporadic forms. Approximately 15% are inherited and associated with coding mutations in the PRNP gene. Acquired prion diseases include iatrogenic CJD, kuru (245300), variant CJD (vCJD) in humans, scrapie in sheep, and bovine spongiform encephalopathy (BSE) in cattle.
Overview Creutzfeldt-Jakob (KROITS-felt YAH-kobe) disease, also known as CJD, is a rare brain disorder that leads to dementia. It belongs to a group of human and animal diseases known as prion disorders. Symptoms of Creutzfeldt-Jakob disease can be similar to those of Alzheimer's disease. But Creutzfeldt-Jakob disease usually gets worse much faster and leads to death. Creutzfeldt-Jakob disease (CJD) received public attention in the 1990s when some people in the United Kingdom became sick with a form of the disease.
A subacute fatal neurodegenerative disease belonging to the group of prion diseases, characterized by a clinical triad of dementia, myoclonus, and EEG anomalies, along with neuropathological evidence of neuronal loss, spongiform changes, and astrocytosis. There are three types of CJD: sporadicCJD (sCJD), inherited CJD , and iatrogenic and variant CJD (vCJD).
Inherited or familial Creutzfeldt-Jakob disease (fCJD) is a very rare form of genetic prion disease (see this term) characterized by typical CJD features (rapidly progressive dementia, personality/behavioral changes, psychiatric disorders, myoclonus, and ataxia) with a genetic cause and sometimes a family history of dementia.
Creutzfeldt-Jakob disease (CJD) is a rare fatal brain disorder that usually occurs later in life and runs a rapid course. In the early stages of the disease, patients may have failing memory, behavior changes, impaired coordination, and vision problems. As CJD progresses, mental deterioration becomes severe, and they can have uncontrolled movements, blindness, weakness, and go into a coma. This condition often leads to death within a few weeks or months after symptoms begin. About 90 percent of patients do not survive for more than one year. In the United States, about 300 people are diagnosed with this condition each year.
Prion disease represents a group of conditions that affect the nervous system in humans and animals. In people, these conditions impair brain function, causing changes in memory, personality, and behavior; a decline in intellectual function (dementia); and abnormal movements, particularly difficulty with coordinating movements (ataxia). The signs and symptoms of prion disease typically begin in adulthood and worsen with time, leading to death within a few months to several years. Frequency These disorders are very rare. Although the exact prevalence of prion disease is unknown, studies suggest that this group of conditions affects about one person per million worldwide each year. Approximately 350 new cases are reported annually in the United States.
A group of human prion diseases characterized by progressive, invariably fatal neurodegeneration resulting from accidental transmission of prions. The group comprises iatrogenic Creutzfeldt-Jakob disease (CJD), which results from transmission of CJD prions in the course of medical procedures or treatments, and variant CJD (transmission via consumption of products from prion-diseased cows or via blood transfusion from an affected individual).
PMC 6227606 . PMID 30496103 . ^ a b c d e f g h i j k l m n o p Bern C (July 2015). ... Retrieved 15 April 2020 . ^ a b c d e f g h i j k l m n o p q Nunes MC, Beaton A, Acquatella H, et al. ... "Diagnosis of Chagas disease". In Marcelo Altcheh J, Freilij H (ed.). Chagas Disease: A Clinical Approach . ... "Gastrointestinal Chagas Disease". In Marcelo Altcheh J, Freilij H (eds.). Chagas Disease: A Clinical Approach . ... "Chagas Disease in Europe". In Marcelo Altcheh J, Freilij H (ed.). Chagas Disease: A Clinical Approach .
Overview Chagas (CHAH-gus) disease is an inflammatory, infectious disease caused by the parasite Trypanosoma cruzi. This parasite is found in the feces of the triatomine (reduviid) bug. This bug is also known as the "kissing bug." Chagas disease is common in South America, Central America and Mexico, the primary home of the triatomine bug. Rare cases of Chagas disease have also been found in the southern United States. Also called American trypanosomiasis, Chagas disease can infect anyone.
A tropical disease mainly found in latin America and transmitted by triatomine insects (mostly Triatoma infestans and Rhodnius prolixus and Panstrongylus megistus ) harboring the hemoflagellate protozoan parasite Trypanosoma cruzi . The disease is characterized by an acute phase which is either asymptomatic or manifest with fever, inflammation at the inoculation site (inoculation chancre or chagoma), unilateral palpebral edema called the Romaña sign (when the triatomine bite occurs near the eye), enlarged lymph nodes, and splenomegaly. The chronic phase is lifelong and development of chagasic cardiomyopathy (30%; complex arrhythmias, heart failure, and thromboembolic events), digestive (10%; megaoesophagus and megacolon), neurological (10%; stroke, peripheral neuropathy and autonomic dysfunction), or mixed alterations (10%) may be observed. These can all lead to high morbidity and mortality rates.
Epithelioid sarcoma Micrograph of an epithelioid sarcoma. H&E stain . Specialty Oncology Epithelioid sarcoma is a rare soft tissue sarcoma arising from mesenchymal tissue and characterized by epithelioid -like features. ... PMID 24376795 . ^ Soft Tissue Sarcoma Staging at eMedicine ^ a b de Visscher, Sebastiaan A. H. J.; van Ginkel, Robbert J.; Wobbes, Theo; Veth, René P. H.; ten Heuvel, Suzanne E.; Suurmeijer, Albert J. H.; Hoekstra, Harad J. (2006). "Epithelioid sarcoma: Still an only surgically curable disease". ... D.; Van Tine, B. A.; Reed, D. R.; Okuno, S. H.; Butrynski, J. E.; Adkins, D. R.; Hendifar, A.
Epithelioid sarcoma (ES) is a rare cancerous tumor that most often occurs in the soft tissue of the fingers, hands and forearms of young adults. It can also occur elsewhere in the body. ES usually begins as a painless, firm growth or bump that may be accompanied by an open wound ( ulceration ) in the skin covering the growth. This type of tumor often comes back after treatment or spreads to other parts the body (metastasis). The cause of epithelioid sarcoma is unknown. It is diagnosed by a clinical examination and by testing a small sample of the tumor (biopsy) in a laboratory. Epithelioid sarcoma is treated with surgery to remove all the cancer cells ( wide local excision ) and sometimes with radiation therapy.
Epithelioid sarcoma is a rare, soft tissue tumor characterized by high incidence of local recurrence, regional lymph node involvement and distant metastases. It commonly affects the soft tissue under the skin of a finger, hand, forearm, lower leg or foot, less often other areas of the body.
A number sign (#) is used with this entry because lymphangioleiomyomatosis (LAM) can occur in association with tuberous sclerosis complex (TSC; 191100) due to mutations in the TSC1 (605284) or TSC2 (191092) genes. Sporadic LAM typically results from 2 somatic mutations in the TSC2 gene, although a fraction of sporadic LAM is caused by germline mutations in the TSC1 gene. Clinical Features Pulmonary lymphangiomyomatosis, also known as pulmonary lymphangioleiomyomatosis (Urban et al., 1999), is a rare disease that occurs almost exclusively in women. It was first described by Van Stossel (1937). The average age at onset of symptoms, which include shortness of breath (67%), lung collapse (25%), coughing (12%), and chest pain (10%), is 33 years (Taylor et al., 1990; Johnson et al., 1993). Chest x-rays typically show a diffuse interstitial infiltrate, with no predominance in any 1 lung zone.
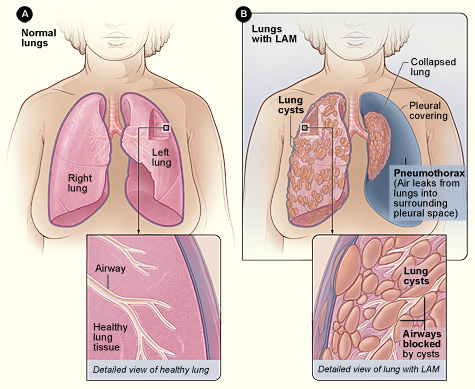
Lymphangioleiomyomatosis (LAM) is a condition that affects the lungs, the kidneys , and the lymphatic system . The lymphatic system consists of a network of vessels that transport lymph fluid and immune cells throughout the body. Lymph fluid helps exchange immune cells, proteins, and other substances between the blood and tissues. LAM is found almost exclusively in women. It often occurs as a feature of an inherited syndrome called tuberous sclerosis complex. When LAM occurs alone it is called isolated or sporadic LAM. Signs and symptoms of LAM most often appear during a woman's thirties.
Lymphangioleiomyomatosis (LAM) is a multiple cystic lung disease characterized by progressive cystic destruction of the lung and lymphatic abnormalities, frequently associated with renal angiomyolipomas (AMLs). LAM occurs either sporadically or as a manifestation of tuberous sclerosis complex (TSC). Epidemiology Sporadic LAM affects around 1/500,000-1/125,000 adult women in Europe. TSC is present in 1 in 6,000 births. Pulmonary LAM is present in up to 30-40% of adult TSC cases. LAM affects almost exclusively females. Clinical description Defining manifestations of the disease are respiratory and include progressive dyspnea, pneumothorax and chylothorax.
Lymphangioleiomyomatosis (lim-FAN-je-o-LI-o-MI-o-ma-TO-sis), or LAM, is a rare cystic lung disease that mostly affects women in their mid-forties. In LAM, an unusual type of cell begins to grow out of control throughout the body, including in the lungs, lymph nodes and vessels, and kidneys. Over time, these LAM cells form cysts and clusters of cells, which grow throughout the lungs and slowly block the airways. They also destroy the normal lung tissue and replace it with cysts. As a result, air cannot move freely in and out of the lungs, and the lungs cannot supply enough oxygen to the body’s other organs. Some people also develop growths called angiomyolipomas (AMLs) in the kidneys.
Molecular analysis detected a homoplasmic 3460G-A mutation in blood and spinal cord. Her mtDNA haplotype H and HLA-DR8 status did not explain the severe phenotype. ... In contrast, the risk of visual failure was slightly decreased (OR = 0.79) when 11778G-A was present on haplotype H. By examining data from a population-based study (1970-2004), Puomila et al. (2007) estimated that the prevalence of LHON in Finland is 1 in 50,000, and that 1 in 9,000 Finns is a carrier of 1 of the 3 LHON primary mutations (MTND4, 11778G-A; MTND1, 3460G-A; and MTND6, 14484T-C).
Summary Clinical characteristics. Leber hereditary optic neuropathy (LHON) is characterized by bilateral, painless, subacute visual failure that develops during young adult life. Males are four to five times more likely than females to be affected. Affected individuals are usually entirely asymptomatic until they develop visual blurring affecting the central visual field in one eye; similar symptoms appear in the other eye an average of two to three months later. In about 25% of cases, visual loss is bilateral at onset. Visual acuity is severely reduced to counting fingers or worse in the majority of cases, and visual field testing shows an enlarging dense central or centrocecal scotoma. After the acute phase, the optic discs become atrophic. Significant improvement in visual acuity is rare and most persons qualify for registration as legally blind (visual acuity ≤20/200).
In 95% of cases worldwide, Leber hereditary optic atrophy (LHON) is due to 1 of 3 point mutations of mitochondrial DNA in genes that code for complex I of the respiratory chain: 3460G-A in MTND1 (516000.0001), 11778G-A in MTND4 (516003.0001), and 14484T-C in MTND6 (516006.0001). That only approximately 50% of male and 10% of female mutation carriers develop symptoms (Newman, 2002) indicates a requirement for additional genetic or environmental factors for phenotypic expression of LHON. Epidemiologic studies failed to substantiate anecdotal reports of a link with excess alcohol and tobacco (Kerrison et al., 2000). Bu and Rotter (1991) concluded that available pedigree data on LHON are most consistent with a 2-locus disorder, with one responsible gene being mitochondrial and the other nuclear and X chromosome-linked. They demonstrated that a proportion of affected females are probably heterozygous at the X chromosome-linked locus and are affected due to unfortunate X chromosome inactivation, thus providing an explanation for the later age of onset in females.
Leber hereditary optic neuropathy (LHON) is a condition characterized by vision loss. Vision loss is typically the only symptom of LHON. Some families with additional signs and symptoms have been reported and are said to have " LHON plus ", a condition which includes vision loss, tremors, and abnormalities of the electrical signals that control the heartbeat ( cardiac conduction defects ). Some affected individuals develop features similar to multiple sclerosis . LHON is caused by mutations in the MT-ND1 , MT-ND4 , MT-ND4L , and MT-ND6 genes. LHON has a mitochondrial pattern of inheritance; however, there are many cases in which there are no other cases of LHON in the family.
Leber hereditary optic neuropathy (LHON) is an inherited form of vision loss. Although this condition usually begins in a person's teens or twenties, rare cases may appear in early childhood or later in adulthood. For unknown reasons, males are affected much more often than females. Blurring and clouding of vision are usually the first symptoms of LHON. These vision problems may begin in one eye or simultaneously in both eyes; if vision loss starts in one eye, the other eye is usually affected within several weeks or months.
Leber's hereditary optic neuropathy (LHON) is a mitochondrial neurodegenerative disease affecting the optic nerve and often characterized by sudden vision loss in young adult carriers. Epidemiology Prevalence of the disease is not well known but is estimated at 1/15,000 - 1/50,000 people worldwide. Clinical description Sudden, painless, acute or subacute central vision loss is often noted between the ages of 18 to 30. It affects both eyes simultaneously or sequentially with vision loss in the second eye occurring weeks to months after the first. Visual loss generally occurs subacutely (over a period of several weeks) and then stabilizes.
PMID 19132029 . ^ Goethe, WH; Bäter, H; Laban, C (October 1989). "Barodontalgia and barotrauma in the human teeth: findings in navy divers, frogmen, and submariners of the Federal Republic of Germany". ... Walheim Shannon Walker John Morgan Wells Joachim Wendler Douglas H. Wheelock Peggy Whitson Dafydd Williams Jeffrey Williams Sunita Williams Gregory R.
. ^ Lundgren CEG, Tjernström O, Ornhagen H (September 1974). "Alternobaric vertigo and hearing disturbances in connection with diving: an epidemiologic study" . ... Walheim Shannon Walker John Morgan Wells Joachim Wendler Douglas H. Wheelock Peggy Whitson Dafydd Williams Jeffrey Williams Sunita Williams Gregory R.
See also [ edit ] Nitrogen narcosis – Reversible narcotic effects of respiratory nitrogen at elevated partial pressures Decompression sickness – Disorder caused by dissolved gases in the tissues forming bubbles during reduction of the surrounding pressure References [ edit ] ^ a b c d e f g h Bennett, Peter B; Rostain, Jean Claude (2003). ... Walheim Shannon Walker John Morgan Wells Joachim Wendler Douglas H. Wheelock Peggy Whitson Dafydd Williams Jeffrey Williams Sunita Williams Gregory R.
PMC 3169834 . PMID 21994818 . ^ COMROE, JULIUS H. (7 July 1945). "OXYGEN TOXICITY". ... Walheim Shannon Walker John Morgan Wells Joachim Wendler Douglas H. Wheelock Peggy Whitson Dafydd Williams Jeffrey Williams Sunita Williams Gregory R.
Taravana is also used to describe someone who is "crazy because of the sea". References [ edit ] ^ Rahn, H.; Yokoyama, T. (1965). Physiology of Breath-Hold Diving and the Ama of Japan . ... Walheim Shannon Walker John Morgan Wells Joachim Wendler Douglas H. Wheelock Peggy Whitson Dafydd Williams Jeffrey Williams Sunita Williams Gregory R.
Science 2004; 304 : 1932-1938. ^ a b Lim WL, Xing H, Wong KH, et al. The lack of an epidemiological link between HIV type 1 infections in Hong Kong and Mainland China.
Continuing Education in Anaesthesia, Critical Care & Pain . 14 (2): 47–51. doi : 10.1093/bjaceaccp/mkt031 . ^ a b c d e f g h Staff. "Laryngospasm" . Heartburn/GERD Guide . ... Walheim Shannon Walker John Morgan Wells Joachim Wendler Douglas H. Wheelock Peggy Whitson Dafydd Williams Jeffrey Williams Sunita Williams Gregory R.