-
Sebaceous Lymphadenoma
Wikipedia
See also [ edit ] Lymph node Lymphoma Salivary gland neoplasm References [ edit ] ^ Mishra, A.; Tripathi, K.; Mohanty, L.; Nayak, M. "Sebaceous lymphadenoma of the parotid gland" .

-
Benign Lymphoepithelial Lesion
Wikipedia
Stone; Arezou Khosroshahi; Vikram Deshpande; John K. C. Chan; J. Godfrey Heathcote; Rob Aalberse; Atsushi Azumi; Donald B. ... Eur J Ophthalmol . 16 (2): 199–203. PMID 16703534 . Tsubota, K; Fujita, H; Tsuzaka, K; Takeuchi, T (Jun 2000). ... Yamamoto, M; Harada, S; Ohara, M; Suzuki, C; Naishiro, Y; Yamamoto, H; Takahashi, H; Imai, K (Feb 2005). "Clinical and pathological differences between Mikulicz's disease and Sjögren's syndrome".
-
Bronchopulmonary Dysplasia
Wikipedia
. ^ Sahni, R; Ammari, A; Suri, MS; Milisavljevic, V; Ohira-Kist, K; Wung, JT; Polin, RA (Jan 2005). "Is the new definition of bronchopulmonary dysplasia more useful?" ... PMID 15538399 . ^ Ehrenkranz, RA; Walsh, MC; Vohr, BR; Jobe, AH; Wright, LL; Fanaroff, AA; Wrage, LA; Poole, K; National Institutes of Child Health and Human Development Neonatal Research, Network (Dec 2005). ... PMID 22564302 . ^ Hayes D, Jr; Wilson, KC; Krivchenia, K; Hawkins, SMM; Balfour-Lynn, IM; Gozal, D; Panitch, HB; Splaingard, ML; Rhein, LM; Kurland, G; Abman, SH; Hoffman, TM; Carroll, CL; Cataletto, ME; Tumin, D; Oren, E; Martin, RJ; Baker, J; Porta, GR; Kaley, D; Gettys, A; Deterding, RR (1 February 2019). ... Bronchopulmonary Dysplasia on National Institutes of Health External links [ edit ] Classification D ICD - 10 : P27.1 ICD - 9-CM : 770.7 MeSH : D001997 DiseasesDB : 1713 External resources MedlinePlus : 001088 eMedicine : ped/289 Patient UK : Bronchopulmonary dysplasia Orphanet : 70589 v t e Conditions originating in the perinatal period / fetal disease Maternal factors complicating pregnancy, labour or delivery placenta Placenta praevia Placental insufficiency Twin-to-twin transfusion syndrome chorion / amnion Chorioamnionitis umbilical cord Umbilical cord prolapse Nuchal cord Single umbilical artery presentation Breech birth Asynclitism Shoulder presentation Growth Small for gestational age / Large for gestational age Preterm birth / Postterm pregnancy Intrauterine growth restriction Birth trauma scalp Cephalohematoma Chignon Caput succedaneum Subgaleal hemorrhage Brachial plexus injury Erb's palsy Klumpke paralysis Affected systems Respiratory Intrauterine hypoxia Infant respiratory distress syndrome Transient tachypnea of the newborn Meconium aspiration syndrome Pleural disease Pneumothorax Pneumomediastinum Wilson–Mikity syndrome Bronchopulmonary dysplasia Cardiovascular Pneumopericardium Persistent fetal circulation Bleeding and hematologic disease Vitamin K deficiency bleeding HDN ABO Anti-Kell Rh c Rh D Rh E Hydrops fetalis Hyperbilirubinemia Kernicterus Neonatal jaundice Velamentous cord insertion Intraventricular hemorrhage Germinal matrix hemorrhage Anemia of prematurity Gastrointestinal Ileus Necrotizing enterocolitis Meconium peritonitis Integument and thermoregulation Erythema toxicum Sclerema neonatorum Nervous system Perinatal asphyxia Periventricular leukomalacia Musculoskeletal Gray baby syndrome muscle tone Congenital hypertonia Congenital hypotonia Infections Vertically transmitted infection Neonatal infection rubella herpes simplex mycoplasma hominis ureaplasma urealyticum Omphalitis Neonatal sepsis Group B streptococcal infection Neonatal conjunctivitis Other Miscarriage Perinatal mortality Stillbirth Infant mortality Neonatal withdrawalIL1B, TXN, POSTN, HIF1A, CAT, EPAS1, EDN1, CASP9, CASP8, CASP3, BID, BAX, CXCL1, TP53, SPOCK2, NBL1, CTNNA3, AGBL1, IL6, BDNF, VEGFA, IL1RAPL1, AGBL1-AS1, TGFB1, DMD, MICOS10-NBL1, ACE, NFE2L2, TNF, ACTB, FGF10, SLC6A4, SFTPB, ANK3, PPARG, ERBB4, OPN1LW, CCN2, COMT, IL10, IL1A, FN1, HMOX1, SFTPA1, SFTPD, AHR, IL1RN, ELN, TIMP1, FGF7, GABPA, MMP9, CYP1A1, GAD1, EPO, GSTT1, GSTM1, NOS3, DISC1, CTNNB1, SFTPC, P2RX7, REN, SOD3, CXCL12, SCGB1A1, CACNA1C, P2RX5, TNFAIP6, IL18, IL17A, TGM2, HPGDS, SGSM3, SELL, TGFA, SLC6A3, CCL2, SOD2, IGF1, IFNG, TNC, TLR4, NRG1, HDAC2, TXNRD1, TPH1, CCL24, KDR, P2RX4, MMP2, P2RX3, P2RX1, NR3C1, P2RY1, P2RY2, PC, PDGFRA, MTHFR, COX2, PDGFRB, POMC, BRD1, MSC, P2RX6, MBL2, LOX, PTGS2, LLGL1, TIMP2, P2RX2, PRPH2, SIRT1, TST, TLR5, ZNF804A, NQO1, MIR34A, NPSR1, FGFR2, MIR17HG, FKBP5, TPH2, ELANE, CRISPLD2, GABRB2, DGKH, DPYSL2, DNMT1, MIR206, SFTPA2, CHRNA7, AKT1, GRK3, CTSG, CRP, CENPJ, CRHR1, P2RX5-TAX1BP3, DDIT3, NLRP3, MIR21, AIMP1, MIR29B2, CLOCK, MIR29B1, ARHGEF7, MIR17, ZBED4, CDK2AP2, IL18BP, MIR30A, PLOD3, MIR219A1, FHL5, MIRLET7C, IPO13, LPAR2, LRPPRC, VAPA, MIR214, SYBU, SYNJ1, SOD2-OT1, WASH6P, SUMO1, MIR421, ZGLP1, MIR876, TRAF1, TAF9BP2, PER2, WASHC1, LINC01672, RN7SL263P, TSPAN8, MTCO2P12, H3P8, VASP, VDR, POTEM, MIR489, MIR431, XBP1, CXCR4, SLC14A2, BDNF-AS, PLF, MIR335, MAD1L1, POTEKP, MDD1, PLA2G10, IL18R1, CCN5, TLR6, GPNMB, CEP85L, IL33, LAMTOR2, MAP1LC3B, LMAN2L, EHMT1, MCPH1, ZDHHC8, MPPE1, F11R, SHANK1, ASCC1, ADIPOR1, RMDN1, PINK1, LEF1, CLDN18, ETNPPL, NPAS3, NIF3L1, MZB1, NRN1, PTBP2, SF3B6, ZNF410, TREM1, DLL4, UGT1A1, ACKR3, DYM, RMDN3, MIXL1, ESAM, MALAT1, BOC, GSTK1, ACTBL2, CACNG2, CHDH, CAP2, PDLIM5, HCA1, CXCR6, PDE10A, SUB1, KANK4, EML5, RAB40B, RIPK3, RMDN2, RPP14, SLCO6A1, ACOT7, SHANK2, SCGB3A2, TIRAP, SYNE1, DDAH2, DDAH1, IL17F, CACNA2D4, EML2, TENM4, INTU, SERPINA3, MAPK3, TLR2, TIMP3, FOXF2, FOXF1, FGFR4, F3, F2RL1, EZH2, ETS1, EPHA1, ENG, SERPINB1, EGFR, EDNRA, LPAR1, DUSP5, DUSP1, DRD4, DDT, FOXM1, FLNA, FPR1, GNAS, GRP, GRIK2, GRN, GPR42, MCHR1, UTS2R, GPC1, GLP1R, G6PD, GFAP, GDF2, GCH1, GCG, GC, GATA6, GAD2, DAG1, CYP2B6, CYP1B1, AGT, ATF4, ARG1, APOE, ANXA1, ANGPT1, ALB, AGTR1, ADRA2B, BMP7, ADRA1A, ADCYAP1, ADCY2, ADA, ACVRL1, ACTG2, ACTG1, BCL2, BMPR2, CSF3, CDKN2A, CRYZ, CRH, CMKLR1, CLU, CHRNA2, CHRM2, CFL1, CDKN1A, BPI, CD44, CD40, CD34, CAMK2A, CALB2, CACNA1E, BRS3, GSK3B, GSTA6P, GSTP1, RELN, SCN8A, SCN4B, S100A9, RARA, PVALB, PTBP1, PRTN3, PRSS2, CCL18, ABCA3, MAPK1, PLOD2, PLG, PLCG1, PLA2G2A, PFN1, CCL13, SELP, PCNA, SST, THY1, THBS1, TH, TAC1, VAMP7, STAT3, SSTR4, SOX4, SIPA1, SOD1, SUMO2, SMARCA4, SLC18A2, SLC18A1, SLC6A2, SLC1A2, PECAM1, SERPINE1, GSTZ1, ID2, KCNQ2, IRAK1, IMPA2, IL6ST, IL6R, IL4, CCN1, HTR2C, KIT, HTR2A, HTR1B, HTR1A, HSP90AA1, HMGB1, HLA-A, HDAC1, KCNQ3, LCT, OTSC1, MPO, NR4A2, CCN3, NOTCH3, NM, NGF, NFKBIA, NFKB1, MMP16, LDHA, KMT2A, MIF, LOXL2, LOXL1, FADS1, LGALS1, LEP, H3P40

-
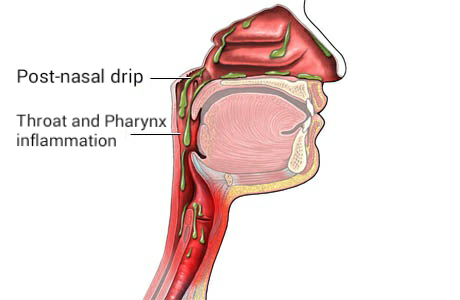
Post-Nasal Drip
Wikipedia
Annals of Allergy, Asthma & Immunology . 106 (4): 267–74, quiz 275. doi : 10.1016/j.anai.2010.09.004 . PMID 21457874 . ^ Kenn K, Balkissoon R (January 2011). "Vocal cord dysfunction: what do we know?" ... S2CID 12436689 . ^ a b c d e f g h i j k l m Flint PW, Haughey BH, Robbins KT, Thomas JR, Niparko JK, Lund VJ, Lesperance MM (2014). ... OCLC 894112159 . ^ a b Rosenfeld RM, Piccirillo JF, Chandrasekhar SS, Brook I, Ashok Kumar K, Kramper M, et al. (April 2015). "Clinical practice guideline (update): adult sinusitis" . ... PMID 16428694 . ^ a b Segboer C, Gevorgyan A, Avdeeva K, Chusakul S, Kanjanaumporn J, Aeumjaturapat S, et al.
-
Astasis
Wikipedia
Clinical Neurology and Neurosurgery . 9 (111): 766–767. doi : 10.1016/j.clineuro.2009.06.003 . ^ Hirayama K, Nakajima M, Kawamura M, Koguchi Y (1994). ... ISBN 978-1-4200-7973-9 . ^ a b Hirayama, K; Nakajima, M; Kawamura, M; Koguchi, Y (1994). ... PMID 12590833 . ^ Kataoka, H; Sigue, K; Kohara, N; Ueno, S (2006). "Novel Representation of Astasia Associated with Posterior Cingulate Infarction" . ... PMID 17516452 . ^ Okun M; Rodriguez R; Foote K; Fernandez Hubert (2007). "The "Chair Test" to Aid in the Diagnosis of Psychogenic Gait Disorders".
-
Poikiloderma Vasculare Atrophicans
Wikipedia
ISBN 978-1-4160-2999-1 . ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad Lambert WC, Everett MA (Oct 1981). ... PMID 7026622 . ^ a b Bonvalet, D.; Colau-Gohm, K.; Belaïch, S.; Civatte, J.; Degos, R. ... PMID 843023 . ^ a b c d Kreuter A, Hoffmamm K, Altmeyer P (Apr 2005). "A case of poikiloderma vasculare atrophicans, a rare variant of cutaneous T-cell lymphoma, responding to extracorporeal photopheresis". ... Mosby Co. pp. 936–941. ^ Sehgal, V. N.; Srivastava, G.; Aggarwal, A. K. (Nov–Dec 2007). "Parapsoriasis: a complex issue".

-
Obsessive–compulsive Spectrum
Wikipedia
Emotions do not affect the behavior but these behaviors are more prevalent in those that suffer with depression. [ citation needed ] Review articles recommend behavioral interventions such as habit reversal training [16] and decoupling . [17] References [ edit ] ^ a b c d e f g h i j k l m McElroy SL; Phillips KA; Keck PE Jr. ... PMID 7961531 . ^ a b Brakoulias, V; Starcevic, V.; Sammut, P.; Berle, D.; Milicevic, D.; Moses, K.; et al. (2011). "Obsessive-compulsive spectrum disorders: a comorbidity and family history perspective". ... PMID 12607216 . Curran S, Matthews K (April 2001). "Response to Yaryura-Tobias et al (2000) negative outcome after neurosurgery for refractory obsessive–compulsive spectrum disorder, World J Biol Psychiatry 1: 197-203". ... "Response to Dr. S. Curran and Dr. K. Matthew's Letter to the editor (World J Biol Psychiatry 2001, 2: 107) concerning Yaryura-Tobias et al (2000) negative outcome after neurosurgery for refractory obsessive–compulsive spectrum disorder, World J Biol Psychiatry 1: 197-203".
-
Leiomyoma
Wikipedia
See also [ edit ] Leiomyosarcoma Elagolix/estradiol/norethindrone acetate References [ edit ] ^ Pedeutour, F.; Quade, B. J.; Sornberger, K.; Tallini, G.; Ligon, A. H.; Weremowicz, S.; Morton, C. ... ISBN 0-7216-2921-0 . ^ Radiologic Pathology Archives: Esophageal Neoplasms: Radiologic-Pathologic Correlation Rachel B. Lewis, Anupamjit K. Mehrotra, Pablo Rodriguez, and Marc S. ... Accessed 2017-07-08 ^ Radiologic Pathology Archives: Esophageal Neoplasms: Radiologic-Pathologic Correlation Rachel B. Lewis, Anupamjit K. Mehrotra, Pablo Rodriguez, and Marc S. ... Archived from the original on 2007-09-27 . Retrieved 2007-03-21 . ^ Patton, K.; Cheng, L.; Papavero, V.; Blum, M.; Yeldandi, A.; Adley, B.; Luan, C.; Diaz, L.; Hui, P.; Yang, X.ESR1, SMAD3, TSC2, SFRP1, WNT5B, INHBA, BCL2, PCNA, PARP1, BAX, EZH2, MKI67, TGFBR1, MYC, HMGA2, PGR, IGF1, IGF2, TP53, CYP19A1, TGFB3, FH, MED12, KIT, FN1, CTNNB1, CDKN2A, VCAN, TGFB1, COMT, AR, FGF2, VEGFA, CYP17A1, ACTB, ESR2, EGF, DES, MMRN1, MMP2, KAT6B, EDN1, CD6, GPER1, CXCL8, CYP1A1, MDM2, CCN1, CYP2B6, GSTM1, DNMT1, EGFR, EGR1, PLAG1, CUX1, TNF, MIR29C, MIR21, MIR200C, KANSL1, ALK, CCND1, PRL, COL1A1, PTEN, CD24, GAPDH, GJA1, PRLHR, PBX3, IGFBP2, PTCH1, FMOD, MUC16, HOXA10, HTC2, CD274, IGF1R, PTGS2, SPIN1, SMAD7, MCL1, SMAD4, RBP1, SMAD2, SHBG, KRAS, TERT, TXN, DLEC1, IL4, YWHAG, IL2, PAEP, IGFBP3, SQSTM1, SERPINE1, H3P10, CD34, ACE, EPO, ALDH1A1, DPT, CDH1, DNMT3A, EWSR1, CCN2, EDNRB, CASP3, CYP1B1, CAV1, SLC27A4, DEPDC5, HDAC6, AKT3, ARNT, MVP, ATF3, NCOR2, ABI2, NCOR1, SLC7A8, ADAMTS4, CHST3, TSIX, EBI3, TNRC6B, RFTN1, SDS, ANGPT2, FST, NCOA2, ATG7, PPIG, KHDRBS1, EBP, NFAT5, NES, WASF3, APEX1, FERMT2, AKAP13, SPINT2, ARTN, KLF4, CA2, XRCC1, XPC, XIST, WNT7A, LAT2, VWF, DST, CALD1, XRCC4, TSC1, TP53BP1, SEC62, TIMP2, TIE1, THBS2, THBS1, XRCC3, BMP2, PTTG1, BECN1, SMUG1, SNURF, BAK1, CCN5, IL18R1, TNFSF9, TNFSF10, NCOA1, NR4A3, TP63, IFITM1, KLF11, RECK, TKTL1, SLC7A5, BCL6, ANGPT1, FGFR3, NUP62, SLC27A1, MIR15B, MIR150, MIRLET7C, MIRLET7A1, SERTM2, CASC15, TBPL2, GADL1, TES, HCAR2, AHR, TET3, PLD5, FAM9A, ADAMTS15, DNAJB7, MIR182, MIR197, AGTR2, AGRP, LOC110806263, MTCO2P12, PGR-AS1, RNA5SP202, TRG-GCC5-1, ACTG1, POTEF, POU5F1P4, POU5F1P3, POTEM, MIR146B, MIR93, PLIN2, MIR29A, MIR221, MAGEC3, OSCP1, RXFP2, ADCY10, ATF7IP, MEG3, ANO1, ANGPT4, BET1L, DCTN4, AKR1B1, SLC27A6, PCDH11X, HPGDS, PDCD4, AGO2, FOXP1, SNORD56, CCND2, EDEM2, HDAC8, CYCSP25, RXFP1, GNRHR2, AZIN2, MED8, NACC1, AKT2, LINC00473, ATAD1, NLRC5, ADGRV1, FERMT3, PDCD1LG2, TET1, NANOG, SNIP1, GGCT, PTPN22, TACR3, CCNG1, ATN1, ITGA2, INSR, DRD1, IL18, CXCR1, DRD2, IL6, EDNRA, TFAP2C, IL1RN, IL1B, IKBKB, IGFBP7, EFNA4, EGR2, IGFBP1, ITGAV, ITGB1, KRT19, LAMB1, MME, DAPK1, MEST, MEN1, DBI, MAP2, DFFA, DFFB, DHCR24, SARDH, LTBP2, LTBP1, CYP4F3, LGALS3, LEP, EPHB2, ERBB2, IFNA13, GRB2, GNRHR, GNRH2, GNRH1, GLI1, FBLN1, GHR, GH1, MSTN, FBN1, FZD2, MTOR, EFEMP1, FGF1, FOXO3, FGFR1, F3, GRIA2, IFNA1, NR3C1, IFI27, TNC, ERBB3, HSPA1B, HSPA1A, HSD17B2, HRAS, HPGD, HOXA11, ERCC2, HMGN2, HIF1A, GTF2H1, ETS1, GRM2, MMP1, MPO, MSH2, SLC5A3, SLC3A2, SLC2A4, SLC2A1, SHH, SFRP4, CDK2, SDHB, CCL2, SATB1, SALL1, S100A11, S100A4, RXRA, TRIM27, REST, SMTN, SMARCB1, PLAAT4, SMN1, CD44, TEK, FGR, TACR1, TAC3, TACR2, TAC1, SYP, STK11, SRC, SPINT1, SOX2, SORD, SNRPN, SMN2, CDKN1A, RAD51B, COX2, CPN1, PECAM1, PDGFRA, PCP4, CPN2, CR2, CSF2, PEBP1, OXTR, OGG1, NTS, NOS3, NGF, NEUROG1, MYLK, CTSL, PGAM1, PIK3CA, PTHLH, PIK3CB, CDKN1B, CEBPB, COL3A1, PSMB9, PRLR, COL4A5, MAPK1, PPP4C, PPARG, POU5F1, PLXNA2, PLP1, COL4A6, PIK3CG, PIK3CD, ACP1

-
Borna Disease
Wikipedia
PMID 2244211 . ^ a b c Rott R, Herzog S, Bechter K, Frese K (1991). "Borna disease, a possible hazard for man?". ... J Affect Disord . 90 (1): 43–7. doi : 10.1016/j.jad.2005.10.008 . PMID 16324750 . ^ Fukuda K, Takahashi K, Iwata Y, et al. (February 2001).
-
Vici Syndrome
Wikipedia
.; Fukushima, Y.; Yamamoto, Y.; Tsunamoto, K.; Nishimura, Y.; Ishida, H.; Toda, T.; Kasubuchi, Y. ... Orphanet Journal of Rare Diseases 11:21 DOI: 10.1186/s13023-016-0399-x ^ Chiyonobu T, Yoshihara T, Fukushima Y, Yamamoto Y, Tsunamoto K et al. (2002) "Sister and brother with Vici syndrome: agenesis of the corpus callosum, albinism, and recurrent infections". ... Nature genetics 45(1): 83-87 ^ Hori I, Otomo T, Nakashima M, Miya F, Negishi Y, Shiraishi H, Nonoda Y, Magara S, Tohyama J, Okamoto N, Kumagai T, Shimoda K, Yukitake Y, Kajikawa D, Morio T, Hattori A, Nakagawa M, Ando N, Nishino I, Kato M, Tsunoda T, Saitsu H, Kanemura Y, Yamasaki M, Kosaki K, Matsumoto N, Yoshimori T, Saitoh S (2017) Defects in autophagosome-lysosome fusion underlie Vici syndrome, a neurodevelopmental disorder with multisystem involvement.

-
Seckel Syndrome
Wikipedia
Harper . [6] [7] See also [ edit ] Koo-Koo the Bird Girl References [ edit ] ^ Harsha Vardhan BG, Muthu MS, Saraswathi K, Koteeswaran D (2007). "Bird-headed dwarf of Seckel" . ... Retrieved 7 January 2011 . ^ Jung M, Rai A, Wang L, Puttmann K, Kukreja K, Koh CJ (2018). "Nephrolithiasis in a 17-Year-Old Male With Seckel Syndrome and Horseshoe Kidneys: Case Report and Review of the Literature".ATR, CENPJ, PCNT, ATRIP, TRAIP, CENPE, PLK4, CEP152, DNA2, LIG4, WDR4, RBBP8, ORC1, ORC4, IGF1, XRCC4, ORC6, DNMT3A, GMNN, CENPF, CDC6, CDT1, LZTR1, KRAS, PTPN11, RAF1, CEP63, SOS1, RIT1, RTTN, NIN, ATM, AIMP2, GRAP2, BRCA1, CD5L, CD69, MCPH1, CHEK1, LARP7, CRK, MAPK14, DONSON, POLDIP2, DNMT1, RNF19A, FANCA, FANCC, PNKP, FAH, H2AX, AHSA1, MAPK1, TELO2, CEP135, TUBGCP6
-
Aa Amyloidosis
Wikipedia
. ^ Chapter 5 in: Mitchell, Richard Sheppard; Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson (2007). Robbins Basic Pathology . ... PMID 23171281 . ^ d'Ythurbide, G; Kerrou, K; Brocheriou, I; Hertig, A (2012). ... Retrieved 12 June 2015 . ^ a b Murakami, T; Ishiguro N; Higuchi K (March 2014). "Transmission of Systemic AA Amyloidosis in Animals" .
-
Granulomatous Meningoencephalitis
Wikipedia
PMID 15521442 . ^ Suzuki M, Uchida K, Morozumi M, Yanai T, Nakayama H, Yamaguchi R, Tateyama S (2003). ... Proceedings of the 50° Congresso Nazionale Multisala SCIVAC . Retrieved 2007-02-18 . ^ Greer, K. A.; Wong, A. K.; Liu, H.; Famula, T.
-
Laboratory Animal Allergy
Wikipedia
. ^ a b Lockey, Richard; Ledford, Dennis K. (2008). "Mammalian Allergens". Allergens and Allergen Immunotherapy . ... Informa Health Care. pp. 201–218. ISBN 978-1-4200-6197-0 . ^ Aoyama K, Ueda A, Manda F, Matsushita T, Ueda T, Yamauchi C (January 1992). ... "Suppression of ouabain-insensitive K-ATPase activity in rabbit nephron segments during chronic hyperkalemia".
-
Hypophosphatemia
Wikipedia
Cardiac monitoring is also advised. [ citation needed ] See also [ edit ] X-linked hypophosphatemia References [ edit ] ^ a b c d e f g h i j k l m n o "Hypophosphatemia" . Merck Manuals Professional Edition . ... Retrieved 23 October 2017 . ^ Shajahan, A., Ajith Kumar, J., Gireesh Kumar, K. P., Sreekrishnan, T. P. and Jismy, K. (2015), Managing hypophosphatemia in critically ill patients: a report on an under-diagnosed electrolyte anomaly.SLC34A1, NR1I2, FGFR1, PHEX, FGF23, SLC34A3, DMP1, FAM20C, VDR, SLC9A3R1, SLC2A2, PTH, PTH1R, ALDOB, CDC73, ALG13, TNFSF11, SUCLG1, GCM2, SNX10, OCRL, TCIRG1, CTNS, CYP27B1, PTCHD1-AS, CASR, AP2S1, GNAS, CLCN5, CYP2R1, CLCN7, COL4A3, FBN1, MEN1, GPT, ENPP1, FGF2, SFRP4, PER2, HRAS, KL, AZI2, TAB3, CORO7, NCKAP1, MEPE, ANKH, TRMO, DTL, GGTLC1, NAA25, SRY, NAMPT, NRAS, BCR, CST3, FGF7, FGFR3, CXCL8, NAP1L1, SLC20A2, ACOT8, SMS, AQP2, TCF21, NAPSA, CLOCK, RAPGEF5, LOC102724197

-
Tropical Eosinophilia
Wikipedia
Reported side effects include headache, fever, pruritus and gastrointestinal upset. [15] The eosinophil count often falls dramatically within 7–10 days of starting treatment. [4] [7] References [ edit ] ^ Jha, Suman K.; Karna, Bibek; Mahajan, Kunal (2020), "Tropical Pulmonary Eosinophilia" , StatPearls , Treasure Island (FL): StatPearls Publishing, PMID 32491456 , retrieved 2020-12-01 ^ a b "Pulmonary Eosinophilia" . ... PMID 17908009 . ^ a b c Boggild, A. K.; Keystone, J. S.; Kain, K. C. (2004).
-
Eales Disease
Wikipedia
Gainesville, Florida: Triad Publishing Company. [ full citation needed ] ^ Deobhakta, Avnish; Chang, Louis K. (2013). "Inflammation in Retinal Vein Occlusion" . ... Retrieved 2019-12-16 . ^ Madhavan, HN; Therese, KL; Doraiswamy, K (March 2002). "Further investigations on the association of Mycobacterium tuberculosis with Eales' disease". ... PMID 23514227 . ^ Comprehensive Ophthalmology, (4th Ed) 2007 by A K Khurana, Professor, Regional Institute of Ophthalmology, Postgraduate Institute of Medical Sciences, Rohtak- 124001, India ^ Eales disease at eMedicine ^ "Eales Disease - an overview | ScienceDirect Topics" . www.sciencedirect.com .
- Metanephric Adenoma Wikipedia
-
Eem Syndrome
Wikipedia
You can help by adding to it . ( July 2017 ) See also [ edit ] Germ layer Integumentary system Hay-Wells syndrome References [ edit ] ^ a b c Hayakawa M, Yanashima K, Kato K, Nakajima A, Yamauchi H (1989). ... External links [ edit ] Classification D ICD - 10 : Q82.4 ICD - 9-CM : 757.31 OMIM : 225280 MeSH : C536190 SNOMED CT : 720856002 External resources Orphanet : 1897 v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation Birthmark

- Fowler's Syndrome Wikipedia








