A rare multiple congenital anomalies/dysmorphic syndrome characterized by a variable combination of dental, cutaneous, ocular, and bone abnormalities, including pyramidal and fused molar roots, taurodontism, an abnormal upper lip without a cupid's bow and thickened and wide philtrum, juvenile glaucoma, syndactyly, and clinodactyly. There have been no further descriptions in the literature since 1973.
Clinical Features Ackerman et al. (1973) described a kindred in which a sister and 2 brothers had pyramidal molar roots. The 2 brothers had juvenile glaucoma and all 6 members of their sibship were said to have an unusual morphology of the upper lip which was full without a Cupid's bow and with a thickened and widened philtrum. Pyramidal, taurodont, or fused molar roots were found in all 3 otherwise unaffected sibs, in both parents, and in sibs, nephews, and nieces of both parents. Were these persons heterozygous for a gene that produced glaucoma and all pyramidal teeth in the 3 sibs who were homozygous? Mouth - Upper lip full - Cupid's bow absent - Philtrum thick and wide Inheritance - Autosomal recessive Eye - Glaucoma, juvenile Teeth - Molar roots pyramidal - Taurodontism ▲ Close
Formation of the line may depend upon the rate of tear secretion. [2] However, the Hudson–Stahli line can be enhanced in hydroxychloroquine toxicity. [3] See also [ edit ] Fleischer ring – corneal iron depositions in keratoconus References [ edit ] ^ Ophthalmology Myron Yanoff, Jay S. Duker Edition 3, illustrated Elsevier Health Sciences, 2008 ISBN 0-323-04332-1 , ISBN 978-0-323-04332-8 ^ Rao, SK; Ananth, VS; Padmanabhan, P (2002-05-01).
Mandibular tori are usually present near the premolars and above the location of the mylohyoid muscle 's attachment to the mandible. [1] In 90% of cases, there is a torus on both the left and right sides.
See also [ edit ] Tamoxifen [4] Raloxifene Triple-negative breast cancer References [ edit ] ^ DeVita, Vincent T.; Lawrence, Theodore S.; Rosenberg, Steven A.; Robert A.
Management is supportive. [ citation needed ] Epidemiology [ edit ] This is a rare disorder with 92 cases reported up to 2017. [1] History [ edit ] This condition was first described in 1975. [3] References [ edit ] ^ a b Luisin M, Chevreau J, Klein C, Naepels P, Demeer B, Mathieu-Dramard M, Jedraszak G, Gondry-Jouet C, Gondry J, Dieux-Coeslier A, Morin G (2017) Prenatal diagnosis of femoral facial syndrome: Three case reports and literature review. Am J Med Genet A ^ Castro S, Peraza E, Zapata M (2014) Prenatal diagnosis of femoral-facial syndrome: case report.
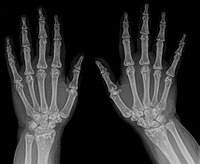
Description Femoral-facial syndrome (FFS), also known as femoral hypoplasia-unusual facies syndrome (FHUFS), is a rare and sporadic multiple congenital anomaly syndrome comprising bilateral femoral hypoplasia and characteristic facial features, such as long philtrum, thin upper lip, micrognathia with or without cleft palate, upward-slanting palpebral fissures, and a short nose with broad tip. Other features, such as renal anomalies, are more variable (summary by Nowaczyk et al., 2010). Clinical Features Daentl et al. (1975) first called attention to a characteristic syndrome of femoral hypoplasia and unusual facies. The facial features include upslanting palpebral fissures, short nose with broad tip, long philtrum, thin upper lip, micrognathia, and cleft palate. Similarities to the caudal regression syndrome were pointed out by Gleiser et al. (1978).
Femoral-facial syndrome is characterized by predominant femoral hypoplasia (bilateral or unilateral) and unusual facies. Epidemiology To date, 55 cases have been reported in the literature. Clinical description Facial features include upslanting palpebral fissures, short nose with broad tip, long philtrum, thin upper lip, micrognathia and cleft palate. The complete syndrome with cleft palate has been reported only in females. The following associated anomalies may be present: vertebral segmentation defects, preaxial polydactyly, ear defects, genitourinary tract abnormalities, lung hypoplasia, dysplastic kidneys, patent arterial duct (see this term). Intellectual development has been reported normal. In two patients, central nervous system anomalies (corticosubcortical atrophy, colpocephaly, partial agenesis of corpus callosum, hypoplasia of the falx cerebri and absent septum pellucidum) have been described.
Femoral facial syndrome (FFS) is a rare condition characterized by underdevelopment of the thigh bone (femoral hypoplasia) and characteristic facial features. Facial features may include upward-slanting eyes, short nose with a broad tip, long space between the nose and upper lip (philtrum), thin upper lip, small lower jaw (micrognathia), and cleft palate. Other features of FFS may include defects of the spinal bones (vertebrae), extra fingers or toes (polydactyly), ear defects, genitourinary abnormalities, underdeveloped lungs, abnormal kidney development, and patent ductus arteriosus . Intellectual development typically is normal. The cause of FFS typically is not known, although genetic factors are thought to play a role. One case has been associated with a chromosome abnormality. Some cases have been reported in association with diabetes in the mother during pregnancy.
In severe cases, when the fever is high enough (generally at or above ~104° F or 40° C), aggressive cooling such as an ice bath and pharmacologic therapy such as benzodiazepines may be deemed appropriate. [1] References [ edit ] ^ Diagnosis and treatment of drug-induced hyperthermia. Musselman, ME. Saely, S. doi: 10.2146/ajhp110543 American Journal of Health-System Pharmacy January 1, 2013 vol. 70 no. 1 34-42 External links [ edit ] Tabor PA (June 1986).
., Dactylonectria spp. and Campylocarpon spp. [5] (cause of black foot disease ) Diplodia seriata (cause of bot canker) [6] Diplodia mutila (cause of Botryosphaeria dieback) Dothiorella iberica Dothiorella viticola Eutypa lata (cause of Eutypa dieback) Fomitiporia mediterranea (cause of esca) Lasiodiplodia theobromae (cause of Botryosphaeria dieback) [6] Neofusicoccum australe Neofusicoccum luteum Neofusicoccom parvum Phaeoacremonium minimum (cause of esca and Petri disease) and other Phaeoacremonium species [5] Phaeomoniella chlamydospora (cause of esca and Petri disease) References [ edit ] ^ Action FA1303 at European Cooperation in Science and Technology (COST) ^ Grapevine Trunk Diseases. symptoms and distribution ( link [ permanent dead link ] ^ Botryosphaeria dothidea associated with grapevine trunk disease in south-eastern Australia. Y. Qiu, S. Savocchia, C. C. Steel and G. J.
Epidemiology [ edit ] Most optic nerve tumors (65 percent) are gliomas that occur somewhere along the anterior visual pathway. [1] See also [ edit ] Melanocytoma References [ edit ] ^ a b c Yanoff, Myron; Duker, Jay S. (2008). Ophthalmology (3rd ed.).
Seminars in Arthritis and Rheumatism . 29 (3): 159–71. doi : 10.1016/S0049-0172(99)80027-4 . PMID 10622680 . MOORE S, CARR AD (January 1952). "Hyperostosis frontalis interna; two contrasting cases".
Clinical Features Knisely et al. (1987) reviewed reported cases of neonatal hemochromatosis, applying rigid criteria as follows: a rapidly progressive clinical course with death in utero or in the early neonatal period; increased tissue iron deposition in multiple sites, particularly in the liver, pancreas, heart, and endocrine glands, with the extrahepatic reticuloendothelial system relatively unaffected; and no evidence for hemolytic disease, syndromes associated with hemosiderosis, or exogenous iron overload from transfusions.
Neonatal hemochromatosis is a disease in which too much iron builds up in the body. This is also called iron overload. Accumulation of iron in the organs is toxic and can cause organ damage. In this form of hemochromatosis, the iron overload begins before birth. This disease tends to progress rapidly and is characterized by liver damage that is apparent at birth or in the first days of life. Babies with the disease may be born very early ( premature ) or struggle to grow in the womb ( intrauterine growth restriction ).
Neonatal hemochromatosis (NH) is an iron storage disorder present at birth. It is a distinct entity that differs from adult hemochromatosis with respect to its molecular origin. Clinical description Clinical signs occur as early as 48 hours after birth and are characterized by the association of severe hepatocellular failure with hyperbilirubinemia, signs of hemorrhage, edema, ascites, hypoglycemia, and lactic acidosis with little to no elevation of transaminases. Etiology The underlying cause of this iron storage disorder is unknown but it may be associated with an anomaly in placental iron transfer. Diagnostic methods Although the diagnosis may be suspected following measurement of transaminase activity, it can only be confirmed by demonstrating the generalized iron overload affecting the salivary glands, liver and pancreas, among other organs.
With incomplete removal, recurrence is common; some surgeons advocate curettage after extraction of teeth to decrease the overall rate of recurrence. [2] See also [ edit ] Cementum Cementogenesis Cementoblast References [ edit ] ^ Sankari Leena S, Ramakrishnan K (2011). "Benign cementoblastoma" .
An acromelic dysplasia that is characterized by severe brachydactyly, peripheral dysostosis with facial dysostosis, nasal hypoplasia, and developmental delay. Epidemiology Less than 80 cases of Acrodysostosis (ACRDYS) have been reported in the literature to date. Clinical description Typical clinical features include severe peripheral dysostosis (short stature and brachydactyly affecting metacarpals, metatarsals and phalanges), facial dysostosis (broad face, widely spaced eyes and maxillonasal hypoplasia), and developmental delay. Advanced skeletal maturation, decreased vertebral interpedicular distance, and obesity are also frequently observed. Several features of acrodysostosis are similar to those present in patients with Albright's hereditary osteodystrophy (AHO) such as short stature, obesity and brachydactyly (in AHO, only 4th and 5th metacarpals and metatarsals).
A number sign (#) is used with this entry because acrodysostosis-1 with or without hormone resistance (ACRDYS1) is caused by heterozygous mutation in the PRKAR1A gene (188830) on chromosome 17q24. Description Acrodysostosis-1 is a form of skeletal dysplasia characterized by short stature, severe brachydactyly, facial dysostosis, and nasal hypoplasia. Affected individuals often have advanced bone age and obesity. Laboratory studies show resistance to multiple hormones, including parathyroid, thyrotropin, calcitonin, growth hormone-releasing hormone, and gonadotropin (summary by Linglart et al., 2011). However, not all patients show endocrine abnormalities (Lee et al., 2012). Genetic Heterogeneity of Acrodysostosis See also ACRDYS2 (614613), caused by mutation in the PDE4D gene (600129) on chromosome 5q12.
A number sign (#) is used with this entry because acrodysostosis-2 with or without hormone resistance (ACRDYS2) is caused by heterozygous mutation in the PDE4D gene (600129) on chromosome 5q12. Description Acrodysostosis-2 is a rare skeletal dysplasia characterized by brachydactyly, facial dysostosis, and spinal stenosis. Many patients have intellectual disability and some have hormone resistance (summary by Michot et al., 2012 and Lee et al., 2012). For a discussion of genetic heterogeneity of acrodysostosis, see ACRDYS1 (101800). Clinical Features Michot et al. (2012) reported 4 unrelated patients, ranging in age from 3 to 7 years, with acrodysostosis-2.
Acrodysostosis refers to a group of genetic disorders of bone growth. Common signs and symptoms include very short fingers and toes, underdeveloped facial bones, a small nose, and short stature. Many individuals with acrodysostosis have developmental delays and intellectual disability. Individuals with acrodysostosis additionally may have hormone resistance, which means that the body does not respond to the certain hormones. There are two types of this disorder, characterized by the presence or absence of hormone resistance and the underlying genetic cause.
Lymphangitis carcinomatosa most often affects people 40–49 years of age. [1] Lymphangitis carcinomatosa may be caused by the following malignancies as suggested by the mnemonic: " C ertain C ancers S pread B y P lugging T he L ymphatics" ( cervical cancer , colon cancer , stomach cancer , breast cancer /bronchiogenic carcinoma, pancreatic cancer , thyroid cancer , laryngeal cancer ) Contents 1 Pathology 2 Prognosis 3 History 4 See also 5 References 6 External links Pathology [ edit ] In most cases, lymphangitis carcinomatosis is caused by the dissemination of a tumor with its cells along the lymphatics. [2] However, in about 20 percent of cases, the inflammation of the lymphatic tubules (lymphangitis) is caused by a tumor that blocks the drainage of the lymph duct.
A number sign (#) is used with this entry because of evidence that familial arrhythmogenic right ventricular dysplasia-1 (ARVD1) is caused by heterozygous mutation in the TGFB3 gene (190230) on chromosome 14q24. Description Arrhythmogenic right ventricular dysplasia (ARVD) is a clinical and pathologic entity for which the diagnosis rests on electrocardiographic and angiographic criteria; pathologic findings, replacement of ventricular myocardium with fatty and fibrous elements, preferentially involve the right ventricular free wall. It is inherited in an autosomal dominant manner with reduced penetrance and is one of the major genetic causes of juvenile sudden death. When the dysplasia is extensive, it may represent the Uhl anomaly ('parchment right ventricle'). The presenting finding is usually recurrent, sustained ventricular tachycardia with left bundle branch block configuration.
Uhl anomaly is characterized by an almost complete absence of the myocardium in the right ventricle resulting in a thin walled nonfunctional right ventricle manifesting with cardiac arrhythmias and right ventricular failure. Cases of partial absence of right ventricular myocardium which remains asymptomatic or mildly symptomatic until adulthood have also been reported. Patients presenting with complete Uhl anomaly should be considered for cardiac transplantation.
Ligneous conjunctivitis is a rare disorder characterized by the buildup of a protein called fibrin which causes inflammation of the conjunctiva (conjunctivitis) and leads to thick, woody (ligneous), inflamed growths that are yellow, white, or red. Ligneous conjunctivitis most often occurs on the inside of the eyelids, but may also affect the sclera, cornea and pupil, leading to vision loss. A systemic form of the condition may occur, affecting the mucous membranes of the larynx, vocal chords, nose, trachea, bronchi, vagina, cervix, and gingiva. The cause of ligneous conjunctivitis is unknown. Autosomal recessive inheritance has been suggested in some cases. Ligneous conjunctivitis is sometimes associated with a condition known as congenital plasminogen deficiency .
CS1 maint: DOI inactive as of January 2021 ( link ) ^ Rubin MA, Bismar TA, Curtis S, Montie JE (July 2004). "Prostate needle biopsy reporting: how are the surgical members of the Society of Urologic Oncology using pathology reports to guide treatment of prostate cancer patients?".