Acta Phytopathologica et Entomologica Hungarica . 52 (2): 157–168. doi : 10.1556/038.52.2017.028 . ISSN 1588-2691 . ^ Pore, R. S. (1986-05-01). "The association of Prototheca spp. with slime flux in Ulmus amencana and other trees".
Local recurrence has been described after simple surgical excision but it is rare. [ citation needed ] Histology [ edit ] Histologically vast array of patterns. Short S-shaped fascicles, inflammation, accelerated mitotic index with normal mitoses.
A rare soft tissue tumor characterized by a solitary mass-forming fibrous proliferation that usually occurs in the subcutaneous tissue, composed of uniform fibroblastic/myofibroblastic cells displaying a loose growth pattern. Upper extremities, trunk, and head and neck are most frequently affected. The lesion typically grows rapidly and almost always measures less than five centimeters in diameter. Macroscopically, it may appear circumscribed or infiltrative but is not encapsulated. Recurrence after excision is very rare, and metastasis does not occur.
Muscular pseudohypertropy - hypothyroidism, also known as Kocher-Debre-Semelaigne syndrome is a rare disorder characterized by pseudohypertrophy of muscles due to longstanding hypothyroidism (see this term). Epidemiology Prevalence is unknown. Clinical description The syndrome usually presents between 18 months and 10 years but has been reported at earlier ages including during the neonatal period. Patients present with clinical features of hypothyroidism, including decreased activity and increased sleep, feeding difficulty and constipation, prolonged jaundice, myxedematous facies, large fontanels (especially posterior), macroglossia, a distended abdomen with umbilical hernia, and hypotonia, along with muscle pseudohypertrophy. Pseudohypertrophy involves the muscles of the extremities, limb girdle, trunk, hands and feet but is more prominent in the limbs, resulting in an athletic appearance. Etiology The etiology of the muscle pseudohypertrophy is not known but it is thought to be a result of long standing hypothyroidism.
Retrieved June 3, 2012 . ^ a b c d e f g Buczacki, S., and Harris, K., Pests, Diseases and Disorders of Garden Plants , Collins, 1998, pp. 481–2. ^ a b c "Damping off" .
. ^ Gallin JI, Fletcher MP, Seligmann BE, Hoffstein S, Cehrs K, Mounessa N (1982). "Human neutrophil-specific granule deficiency: a model to assess the role of neutrophil-specific granules in the evolution of the inflammatory response" .
A number sign (#) is used with this entry because of evidence that specific granule deficiency-1 (SGD1) is caused by homozygous mutation in the CEBPE gene (600749) on chromosome 14q11. Genetic Heterogeneity of Specific Granule Deficiency See also SGD2 (617475), caused by mutation in the SMARCD2 gene (601736) on chromosome 17q23. Clinical Features In mammals, neutrophils contain 2 principal types of granules. The first type, azurophil granules, appear early in neutrophil development and contain lysosomal enzymes, lysozyme (LYZ; 153450), and myeloperoxidase (MPO; 606989). The second type, specific granules, are formed later, lack MPO and hydrolases, but contain lactoferrin (LF; 150210) and the remainder of the cell's complement of lysozyme.
A number sign (#) is used with this entry because of evidence that specific granule deficiency-2 (SGD2) is caused by homozygous mutation in the SMARCD2 gene (601736) on chromosome 17q23. Description Specific granule deficiency-2 is an autosomal recessive immunologic disorder characterized by recurrent infections due to defective neutrophil development. Bone marrow findings include hypercellularity, abnormal megakaryocytes, and features of progressive myelofibrosis with blasts. The disorder is apparent from infancy, and most patients die in early childhood unless they undergo hematopoietic stem cell transplantation. Some patients may have additional findings, including delayed development, mild dysmorphic features, and distal skeletal anomalies (summary by Witzel et al., 2017).
A rare functional neutrophil defect characterized by infantile onset of increased susceptibility to pyogenic infections, especially of the skin, ears, lung, and lymph nodes, with neutrophils lacking specific granules and exhibiting bilobed nuclei on peripheral blood smear. Bone marrow biopsy shows hypercellularity, paucity of neutrophil granulocytes, and progressive myelodysplasia. Additional manifestations may include mild to moderate developmental delay, mild facial dysmorphic features (such as dysplastic ears), and anomalies of bones, teeth, and nails.
Paraphilia Candaules, King of Lydia, Shews his Wife by Stealth to Gyges, One of his Ministers, as She Goes to Bed by William Etty . This image illustrates Herodotus 's version of the tale of Gyges. Duke of Orléans showing his Lover ( Mariette d'Enghien ) by Eugène Delacroix .
. ^ a b Barber JC, Maloney VK, Huang S, et al. (January 2008). "8p23.1 duplication syndrome; a novel genomic condition with unexpected complexity revealed by array CGH" .
8p23.1 duplication syndrome is a rare chromosomal anomaly syndrome, resulting from the partial duplication of the short arm of chromosome 8, with a highly variable phenotype, principally characterized by mild to moderate developmental delay, intellectual disability, mild facial dysmorphism (incl. prominent forehead, arched eyebrows, broad nasal bridge, upturned nares, cleft lip and/or palate) and congenital cardiac anomalies (e.g., atrioventricular septal defect). Other reported features include macrocephaly, behavioral abnormalities (e.g., attention deficit disorder), seizures, hypotonia and ocular and digital anomalies (poly/syndactyly).
A number sign (#) is used with this entry because of evidence that Sneddon syndrome (SNDNS) is caused by compound heterozygous mutation in the CECR1 gene (ADA2; 607575) on chromosome 22q11. One such family has been reported. Mutation in the ADA2 gene can also cause vasculitis, autoinflammation, immunodeficiency, and hematologic defects syndrome (VAIHS; 615688), which shows earlier onset. Description Sneddon syndrome is a noninflammatory arteriopathy characterized by onset of livedo reticularis in the second decade and onset of cerebrovascular disease in early adulthood (summary by Bras et al., 2014). Livedo reticularis occurs also with polyarteritis nodosa, systemic lupus erythematosus, and central thrombocythemia, any one of which may be accompanied by cerebrovascular accidents (Bruyn et al., 1987). Clinical Features Sneddon (1965) described 6 patients (5 females, 1 male), varying in age from 20 to 48 years, who had association of livedo reticularis with cerebrovascular accident.
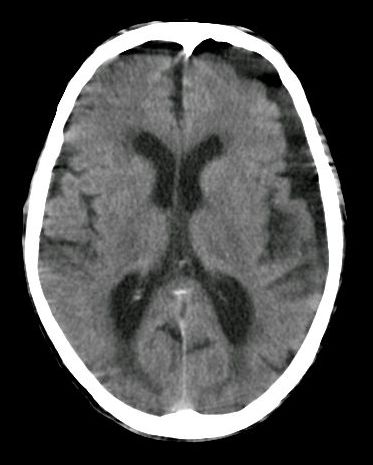
Sneddon syndrome is a rare, progressive condition that affects blood vessels. It is primarily characterized by livedo reticularis (net-like patterns of discoloration on the skin) and neurological abnormalities. Symptoms may include transient ischemic attacks (mini-strokes) and strokes; headache; dizziness; high blood pressure; and heart disease. Reduced blood flow to the brain may cause reduced intellectual ability, memory loss, personality changes, and/or other neurological symptoms. The cause of Sneddon syndrome is often unknown, but it is sometimes associated with an autoimmune disease.
Adenosine deaminase 2 (ADA2) deficiency is a disorder characterized by abnormal inflammation of various tissues. Signs and symptoms can begin anytime from early childhood to adulthood. The severity of the disorder also varies, even among affected individuals in the same family. Inflammation is a normal immune system response to injury and foreign invaders (such as bacteria). However, ADA2 deficiency causes abnormal, unprovoked inflammation that can damage the body's tissues and organs, particularly blood vessels.
Sneddon's syndrome (SS) is a rare non-inflammatory thrombotic vasculopathy characterized by the combination of cerebrovascular disease with livedo racemosa. Epidemiology SS has an estimated annual incidence of approximately 1/250,000. The disease predominantly affects women in young adulthood. Clinical description The mean age of onset of neurological symptoms is 39 years, though the livedo is generally observed up to 10 years earlier and sometimes since childhood. Livedo racemosa is a persistent net-like violaceous-cyanotic, mottled discoloration of the skin affecting primarily the legs and arms, but also involving the buttocks and the trunk, and that is exacerbated by cold or pregnancy. Livedo reticularis, that is limited to the extremities and is visible only in the cold, has also been observed in some cases.
ISBN 978-1-4160-2999-1 . ^ Garavelli L; Pedori S; Zanacca C; et al. (April 2005). "Albright's hereditary osteodystrophy (pseudohypoparathyroidism type Ia): clinical case with a novel mutation of GNAS1".
Mariot et al. (2008) noted that features of AHO may rarely be observed in patients with PHP1b who have decreased expression of G-alpha-s due to epimutations at the GNAS locus. ... Further studies were consistent with a single defect causing deficient hormone receptor-nucleotide complex formation and adenylate cyclase activity in PHP Ia, whereas the biochemical lesion(s) appeared not to affect the complex formation in PHP Ib. ... Gs-alpha transcripts are imprinted in pituitary somatotrophs that secrete growth hormone (Hayward et al., 2001), leading Germain-Lee et al. (2003) to hypothesize that maternally inherited GNAS1 mutations would impair GH secretion through disruption of mediation of the pituitary response to GHRH by G-alpha-s. They studied GH status in 13 subjects with PHP type Ia.
Pseudohypoparathyroidism type 1A (PHP1a) is a type of pseudohypoparathyroidism (PHP; see this term) characterized by renal resistance to parathyroid hormone (PTH), resulting in hypocalcemia, hyperphosphatemia, and elevated PTH; resistance to other hormones including thydroid stimulating hormone (TSH), gonadotropins and growth-hormone-releasing hormone (GHRH); and a constellation of clinical features known as Albright hereditary osteodystrophy (AHO; see this term). Epidemiology Prevalence of PHP (1a and 1b) has been estimated at 1/295,000 in a Japanese study. The prevalence of PHP (1a, 1b and pseudopseudohypoparathyroidism (PPHP); see these terms) has been estimated at 1/150,000in Italy. Clinical description Onset of endocrine symptoms occurs during childhood, although cases with severe hypothyroidism at neonatal screening have been reported. Patients present with varying degrees of AHO features (including obesity).