Contents 1 Asian Games 1.1 1974 Tehran 1.2 1994 Hiroshima 1.3 1998 Bangkok 1.4 2002 Busan 1.5 2006 Doha 1.6 2010 Guangzhou 1.7 2014 Incheon 1.8 2018 Jakarta–Palembang 2 See also 3 References Asian Games [ edit ] 1974 Tehran [ edit ] Main article: 1974 Asian Games Name NOC Sport Banned substance Medals Ref Oh Han-nam South Korea Volleyball (Men) [1] Masushi Ouchi Japan Weightlifting Stimulant (Men's 90 kg) (Men's snatch 90 kg) (Men's clean & jerk 90 kg) [2] Kim Joong-iI North Korea Weightlifting Stimulant (Men's 110 kg) (Men's snatch 110 kg) (Men's clean & jerk 110 kg) [3] 1994 Hiroshima [ edit ] Main article: 1994 Asian Games Name NOC Sport Banned substance Medals Ref Han Qing China Athletics Dihydrotestosterone (Women's 400 m hurdles) [4] Zhang Lei China Canoeing Dihydrotestosterone (Men's C-1 500 m) (Men's C-1 1000 m) (Men's C-2 500 m) [4] Qiu Suoren China Canoeing Dihydrotestosterone (Men's C-2 1000 m) [4] Wang Yan China Cycling Dihydrotestosterone (Women's sprint) [4] Sirisak Kadalee Thailand Football Stimulant [5] Fu Yong China Swimming Dihydrotestosterone (Men's 400 m individual medley) [6] Hu Bin China Swimming Dihydrotestosterone (Men's 50 m freestyle) [6] Lü Bin China Swimming Dihydrotestosterone (Women's 50 m freestyle) (Women's 200 m freestyle) (Women's 200 m individual medley) (Women's 4 × 100 m freestyle relay) (Women's 100 m freestyle) (Women's 100 m backstroke) [6] Xiong Guoming China Swimming Dihydrotestosterone (Men's 200 m freestyle) (Men's 200 m individual medley) (Men's 400 m individual medley) (Men's 4 × 200 m freestyle relay) (Men's 4 × 100 m freestyle relay) [6] Yang Aihua China Swimming Dihydrotestosterone (Women's 400 m freestyle) [6] Zhang Bin China Swimming Dihydrotestosterone (Men's 200 m butterfly) [6] Zhou Guanbin China Swimming Dihydrotestosterone (Women's 400 m freestyle) (Women's 800 m freestyle) [6] 1998 Bangkok [ edit ] Main article: 1998 Asian Games Name NOC Sport Banned substance Medals Ref Abdullah Sabt Ghulam United Arab Emirates Athletics Ephedrine [7] Fakhruddin Abdulmajid United Arab Emirates Karate Ephedrine (Men's kumite 75 kg) [7] Ayed Khawaldeh Jordan Weightlifting Triamterene [8] Jaber Al-Ajmi Kuwait Weightlifting Nandrolone [8] 2002 Busan [ edit ] Main article: 2002 Asian Games Name NOC Sport Banned substance Medals Ref Youssef El-Zein Lebanon Bodybuilding Missed the test ( Men's +90 kg ) [9] 2006 Doha [ edit ] Main article: 2006 Asian Games Name NOC Sport Banned substance Medals Ref Santhi Soundarajan India Athletics Male hormone ( Women's 800 m ) [10] Sayed Faisal Husain Bahrain Bodybuilding ( Men's 70 kg ) [11] Faez Abdul-Hassan Iraq Bodybuilding Nandrolone [12] Kim Myong-hun South Korea Bodybuilding ( Men's 90 kg ) [13] Salem Ghanem Al-Shamsi United Arab Emirates Bodybuilding [14] Kyi Kyi Than Myanmar Weightlifting Diuretic [12] Mya Sanda Oo Myanmar Weightlifting Metabolite ( Women's 75 kg ) [12] Elmira Ramileva Uzbekistan Weightlifting Stanozolol [12] Aleksandr Urinov Uzbekistan Weightlifting Cannabis [12] 2010 Guangzhou [ edit ] Main article: 2010 Asian Games Name NOC Sport Banned substance Medals Ref Suresh Sathya India Athletics Nandrolone [15] Ahmed Dheeb Qatar Athletics Testosterone ( Men's discus throw ) [16] Abdelnasser Awajna Palestine Athletics Norandrosterone [16] Masoud Rigi Iran Boxing Nandrolone [17] Shokir Muminov Uzbekistan Judo Methylhexanamine ( Men's 81 kg ) [18] Jakhongir Muminov Uzbekistan Wrestling Methylhexanamine [19] 2014 Incheon [ edit ] Main article: 2014 Asian Games Name NOC Sport Banned substance Medals Ref Betlhem Desalegn United Arab Emirates Athletics Biological passport abnormalities [20] Khurshed Beknazarov Tajikistan Football Methylhexanamine [21] Nouraddin Al-Kurdi Syria Karate Clenbuterol [22] Yi Sophany Cambodia Soft tennis Sibutramine [23] Park Tae-hwan South Korea Swimming Nebido ( Men's 100 m freestyle ) ( Men's 200 m freestyle ) ( Men's 400 m freestyle ) ( Men's 4 × 100 m freestyle relay ) ( Men's 4 × 200 m freestyle relay ) ( Men's 4 × 100 m medley relay ) [24] Mohammed Jassim Iraq Weightlifting Etiocholanolone [22] Tai Cheau Xuen Malaysia Wushu Sibutramine ( Women's nanquan ) [25] 2018 Jakarta–Palembang [ edit ] Main article: 2018 Asian Games Name NOC Sport Banned substance Medals Ref Kemi Adekoya Bahrain Athletics Stanozolol ( Women's 400 m hurdles ) ( Mixed 4 × 400 m relay ) [26] Sanjivani Jadhav India Athletics Probenecid [26] Nirmala Sheoran India Athletics Drostanolone and Metenolone [27] Kumush Yuldashova Uzbekistan Kurash Stanozolol ( Women's 78 kg ) [28] Pürevdorjiin Orkhon Mongolia Wrestling Stanozolol ( Women's freestyle 62 kg ) [29] Rüstem Nazarow Turkmenistan Wrestling Furosemide [30] See also [ edit ] Asia portal Sports portal Doping at the Commonwealth Games Doping at the Olympics References [ edit ] ^ "South Korea's volleyball silver in the balance" .
3-M syndrome is a disorder that causes skeletal abnormalities including short stature (dwarfism) and unusual facial features. ... In other people with this disorder, the head has an unusually long and narrow shape (dolichocephaly). Intelligence is unaffected by 3-M syndrome, and life expectancy is generally normal. ... Frequency The prevalence of 3-M syndrome is unknown. About 100 individuals worldwide with this disorder have been described in the medical literature. Causes Mutations in the CUL7 gene cause 3-M syndrome in more than three-quarters of affected individuals, including those in the Yakut population. ... However, the specific relationship between CUL7 and OBSL1 gene mutations and the signs and symptoms of 3-M syndrome are unknown. Learn more about the genes associated with 3-M syndrome CUL7 OBSL1 Additional Information from NCBI Gene: CCDC8 Inheritance Pattern This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have mutations.
Males with three M syndrome have hypogonadism and occasionally hypospadias. ... The most striking feature of three M syndrome is the severe intrauterine growth restriction. ... Hypospadias has been seen in a few males with three M syndrome. Note: Female gonadal function appears normal. ... Three M syndrome may also be referred to as Le Merrer syndrome or Yakut short stature syndrome. ... Disorders to Consider in the Differential Diagnosis of Three M Syndrome View in own window Disorder Gene(s) MOI Clinical Features of This Disorder Overlapping w/3-M syndrome Distinguishing from 3-M syndrome Silver-Russell syndrome (SRS) See footnote 1 Simplex IUGR, postnatal growth deficiency SRS often shows limb length asymmetry.
A rare primordial growth disorder characterized by low birth weight, reduced birth length, severe postnatal growth restriction, large head size, a spectrum of minor anomalies (including facial dysmorphism) and normal intelligence. Epidemiology Approximately 200 cases have been reported to date and the condition is rare. However, the phenotype is likely under-recognized. Clinical description Infants present with severe prenatal and postnatal growth retardation, birth weight usually at or below the 2nd centile. Growth impairment is greater in those with CUL7 variants. The head circumference is relatively large. Characteristic facial features include: triangular-shaped face, pointed chin, frontal bossing, hypoplastic midface, fleshy, upturned nose , prominent mouth and lips.
ISBN 9780199934522 – via Oxford Medicine Online. ^ a b c d e f g h i j k "3-M syndrome" . Genetics Home Reference . ... Retrieved November 5, 2019 . ^ a b c d e f g h i j k l m "Three M Syndrome" . DoveMed . November 8, 2016 . ... Retrieved November 5, 2019 . ^ a b c d e f g h i "Three M Syndrome" . Rare Disease Database . ... PMC 2986175 . PMID 19225462 . ^ a b "Three M syndrome 1" . Genetic Testing Registry . ... "Mutations in CUL7, OBSL1 and CCDC8 in 3-M syndrome lead to disordered growth factor signalling" .
The myoclonic jerks typical of SGCE -M-D most often affect the neck, trunk, and upper limbs with less common involvement of the legs. ... Most individuals with SGCE -M-D inherited the disorder from a heterozygous parent who may or may not have clinical signs of M-D (as phenotypic expression in the parent would depend on the gender of the transmitting grandparent). Each child of an individual with SGCE -M-D has a 50% chance of inheriting the pathogenic variant. ... The M-D phenotype of two other families may also be linked to this chromosome region [Schüle et al 2004]. The overall contribution of this locus to M-D cannot be determined until the gene is identified.
Description Microtia-anotia (M-A) can occur either as an isolated defect or in association with other defects. ... M-A also occurs as part of seemingly nonrandom patterns of multiple defects, such as Goldenhar syndrome (164210) (Mastroiacovo et al., 1995). ... Among 1,173,794 births, they identified 172 with M-A, a rate of 1.46/10,000; 38 of the 172 infants (22.1%) had anotia. ... Among the MMI, only holoprosencephaly was preferentially associated with M-A; 4 cases were observed versus 0.7 expected (p = 0.005). ... Mothers with parity 1 had a higher risk of giving birth to an MMI with M-A. Mothers with insulin-dependent diabetes were at significantly higher risk for having a child with M-A.
A congenital malformation of the external ear and the most extreme form of microtia characterized by the complete absence of the external ear and auditory canal, conductive hearing loss, attention deficit disorders and delayed language development.
Anotia Illustration of anotia Specialty Medical genetics Anotia ("no ear") describes a rare congenital deformity that involves the complete absence of the pinna , the outer projected portion of the ear, and narrowing or absence of the ear canal . This contrasts with microtia , in which a small part of the pinna is present. Anotia and microtia may occur unilaterally (only one ear affected) or bilaterally (both ears affected). This deformity results in conductive hearing loss , deafness . Contents 1 Ear development 1.1 Processing sound 1.2 Defects of the ear: anotia/microtia 2 Related syndromes 3 Treatment 4 References 5 External links Ear development [ edit ] This section may be too technical for most readers to understand . Please help improve it to make it understandable to non-experts , without removing the technical details. ( May 2014 ) ( Learn how and when to remove this template message ) Ear development begins in about the third week of human embryonic development.
Agents/circumstances to avoid : Aminoglycosides and noise exposure, especially in those with normal hearing who have the m.1555A>G or m.1494C>T MT-RNR1 pathogenic variants. ... In individuals with hearing loss following aminoglycoside exposure, molecular testing for the pathogenic variants m.1555A>G and m.1494C>T in MT-RNR1 and m.7445A>C/T/G in MT-TS1 can be done first. ... Individuals who have both the m.7444G>A and m.1555A>G ( MT-RNR1 ) pathogenic variants [Pandya et al 2004] or the m.7443A>G pathogenic variant alone do not have skin findings. ... Variants m.961T>G and m.961_962delTinsC(n) have been associated with SNHL [Guaran et al 2013] but may be either benign or low-penetrance pathogenic. ... Maternally inherited diabetes mellitus and deafness (MIDD) is also caused by the MT-TK pathogenic variant m.8296A>G [Kameoka et al 1998], MT-TE pathogenic variants m.14709T>C and m.14692A>G [Rigoli et al 2001, Wang et al 2016], and MT-TG pathogenic variant m.10003T>C [Liu et al 2015].
This procedure has been shown to be beneficial in 50% of cases. [13] [14] Epidemiology [ edit ] This syndrome is predominantly found in young women, but also occurs in children, teenagers and octogenarians. [15] References [ edit ] ^ Van Assen, T.; Brouns, J. A. G. M.; Scheltinga, M. R.; Roumen, R. M. (2015). ... PMID 12003414 . ^ Van Assen, T; De Jager-Kievit, J. W.; Scheltinga, M. R.; Roumen, R. M. (2013). "Chronic abdominal wall pain misdiagnosed as functional abdominal pain" . ... PMID 25444218 . ^ Boelens, O. B.; Scheltinga, M. R.; Houterman, S; Roumen, R. M. (2011). ... PMID 21881494 . ^ Boelens, O. B.; Scheltinga, M. R.; Houterman, S; Roumen, R. M. (2013). ... B.; Van Eerten, P. V.; Scheltinga, M. R.; Roumen, R. M. (2014). "Surgical options after a failed neurectomy in anterior cutaneous nerve entrapment syndrome".
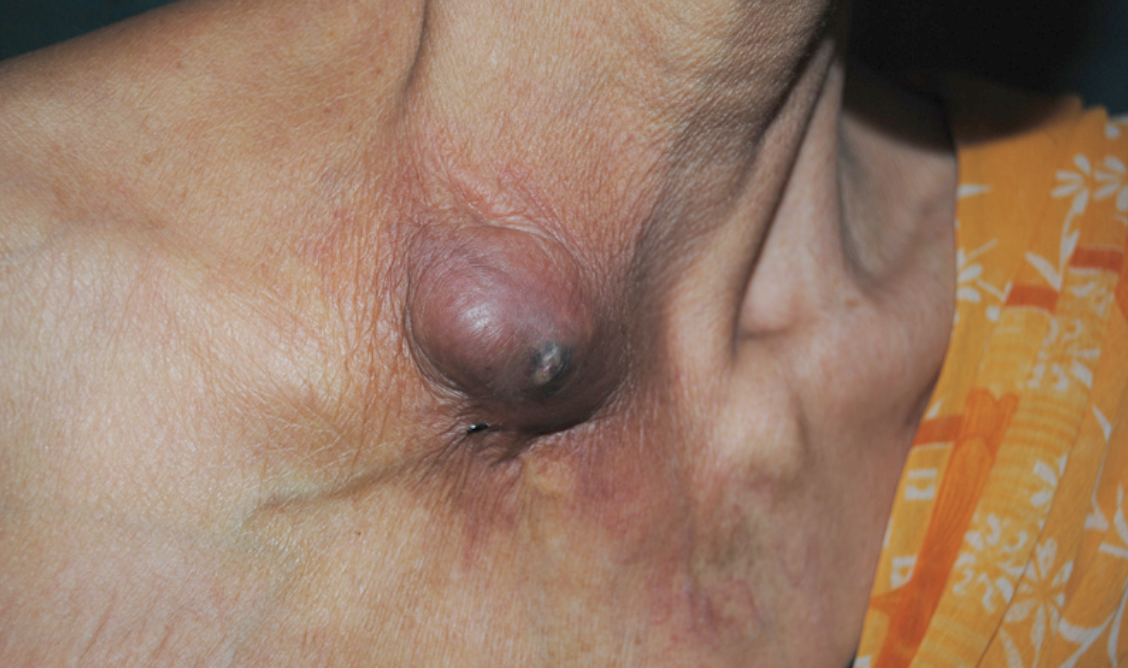
A chronic neuropathic pain syndrome of the abdominal wall caused by entrapment of anterior cutaneous branches of 7 to 12th intercostal nerves along the lateral border of the anterior rectus abdominis fascia causing severe pain and tenderness of the involved dermatome. Epidemiology Around 20% of patients with chronic abdominal pain have abdominal wall pain, which is often caused by ACNES. The prevalence in patients alleged to have functional abdominal pain is estimated to be 3-4%. One out of 50 patients consulting an emergency department for abdominal pain suffers from ACNES. The incidence in general population is probably 1: 4000 to 5000. ACNES, hence may be not so rare as is thought.
PMID 22473397 . ^ Toro, J.; Nickerson, M.; Wei, M.; Warren, M.; Glenn, G.; Turner, M.; Stewart, L.; Duray, P.; Tourre, O.; Sharma, N.; Choyke, P.; Stratton, P.; Merino, M.; Walther, M. M.; Linehan, W. M.; Schmidt, L. S.; Zbar, B. (2003). ... A.; Adam, M. P.; Bird, T. D.; Dolan, C. R.; Fong, C. ... A.; Mikhaĭlov, O. I.; Sekamova, S. M.; Kornev, B. M.; Osipova, I. N.; Osipenko, V. ... PMID 17476294 . ^ Matyakhina, L.; Freedman, R. J.; Bourdeau, I.; Wei, M. H.; Stergiopoulos, S. G.; Chidakel, A.; Walther, M.; Abu-Asab, M.; Tsokos, M.; Keil, M.; Toro, J.; Linehan, W.
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is a condition that causes benign tumors of smooth muscle tissue in the skin (cutaneous leiomyomas) and in the uterus in females (uterine leiomyomas, or fibroids). The condition also increases the risk of kidney cancer. Signs and symptoms usually begin in adulthood as skin growths appear on the torso, arms, legs, and occasionally on the face. They tend to increase in size and number over time. About 10% to 16% of people with HLRCC develop a type of kidney cancer called renal cell cancer; symptoms of this cancer may include lower back pain, blood in the urine, and/or a mass in the kidney that can be felt by a physician. Some people have no symptoms until the cancer is advanced. HLRCC is caused by mutations in the FH gene and is inherited in an autosomal dominant manner.
Summary Clinical characteristics. FH tumor predisposition syndrome is characterized by cutaneous leiomyomata, uterine leiomyomata (fibroids), and/or renal tumors. Pheochromocytoma and paraganglioma have also been described in a small number of families. Cutaneous leiomyomata appear as skin-colored to light brown papules or nodules distributed over the trunk and extremities, and occasionally on the face, and appear at a mean age of 30 years, increasing in size and number with age. Uterine leiomyomata tend to be numerous and large; age at diagnosis ranges from 18 to 53 years, with most women experiencing irregular or heavy menstruation and pelvic pain. Renal tumors are usually unilateral, solitary, and aggressive. They are associated with poor survival due to clinical aggressiveness and propensity to metastasize despite small primary tumor size.
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is a disorder in which affected individuals tend to develop benign tumors containing smooth muscle tissue (leiomyomas) in the skin and, in females, the uterus . This condition also increases the risk of kidney cancer. In this disorder , growths on the skin (cutaneous leiomyomas) typically develop in the third decade of life. Most of these growths arise from the tiny muscles around the hair follicles that cause "goosebumps". They appear as bumps or nodules on the trunk, arms, legs, and occasionally on the face. Cutaneous leiomyomas may be the same color as the surrounding skin, or they may be darker.
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is a hereditary cancer syndrome characterized by a predisposition to cutaneous and uterine leiomyomas and, in some families, to renal cell cancer. Epidemiology The prevalence is unknown. Over 200 families with HLRCC have been reported. Clinical description Disease onset can occur at any age, but is more common in young adults and elderly patients. Multiple or single benign cutaneous leiomyomas are common and usually present at around the age of 25 (range from 10-47 years) as firm papules or nodules that are skin colored to light brown. They are usually localized to the trunk and extremities but sometimes on the face.
A number sign (#) is used with this entry because multiple cutaneous and uterine leiomyomatosis with or without renal cell carcinoma, also referred to as hereditary leiomyatosis and renal cell cancer (HLRCC), is caused by heterozygous mutation in the gene encoding fumarate hydratase (FH; 136850) on chromosome 1q43. Homozygous mutation in the FH gene causes fumarase deficiency (FMRD; 606812). Description Hereditary leiomyomatosis and renal cell cancer is an autosomal dominant tumor predisposition syndrome characterized by the variable development of 3 tumors: cutaneous piloleiomyomata that develop in essentially all patients by age 40 years; leiomyomata (fibroids) of the uterus, and rarely leiomyosarcomas, at a mean age of 30 years (range, 18 to 52 years); and type 2 papillary renal cell carcinoma at a mean age of 46 years (range, 17 to 75 years), which occurs in about 20% of patients. Type 2 papillary renal cell carcinoma is a pathologic subtype characterized by large tumor cells with eosinophilic cytoplasm and pseudostratified nuclei; it shows an aggressive clinical course. Some patients with FH mutations may develop collecting duct renal cell carcinoma.
You can help by adding to it . ( August 2017 ) References [ edit ] ^ Le Merrer M, David A, Goutieres F, Briard ML (October 1992). ... C.; Kim, C.; Hori, M.; Powell, B. R.; Stewart, F.; Félix, T. M. M.; Van Den Ende, J.; Wisniewska, M.; Kayserili, H. L.; Rump, P.; Nampoothiri, S.; Aftimos, S.; Mey, A.; Nair, L. D. V.; Begleiter, M. L.; De Bie, I.; Meenakshi, G.; Murray, M. L.; Repetto, G. M.; Golabi, M.; Blair, E.; Male, A.; Giuliano, F.; Kariminejad, A.; Newman, W.
A number sign (#) is used with this entry because deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures syndrome (DOORS) is caused by homozygous or compound heterozygous mutation in the TBC1D24 gene (613577) on chromosome 16p13. Description The DOOR syndrome is an acronym for deafness, onychodystrophy, osteodystrophy, and mental retardation. Cantwell (1975) suggested this designation for the disorder, which can also include triphalangeal thumbs, seizures, and abnormal dermatoglyphics. Inheritance is autosomal recessive. See also DDOD syndrome (124480), which shows autosomal dominant inheritance of congenital deafness and onychodystrophy without mental retardation. Clinical Features Walbaum et al. (1970) described a brother and sister with mental retardation, perceptive deafness, dysplasia of the fingernails, triphalangeal thumbs, hypoplasia of the terminal phalanges, and 'decapsalidic' fingerprints, i.e., an arch pattern on each finger.
Deafness onychodystrophy osteodystrophy and mental retardation (DOOR) syndrome is a rare genetic disorder that is usually recognized shortly after birth. The term DOOR is an acronym with each letter representing a common feature in affected individuals: (D) eafness due to a defect of the inner ear or auditory nerve; (O)nychodystrophy or malformation of the nails; (O)steodystrophy, meaning malformation of certain bones; and mild to profound intellectual disability (represented by the "R"). In some cases, individuals may also experience seizures. This condition is inherited in an autosomal recessive fashion; the exact genetic cause is unknown.
A rare multiple congenital anomalies-intellectual disability syndrome characterized by sensorineural hearing loss (deafness), onychodystrophy, osteodystrophy, mild to profound intellectual disability, and seizures. Epidemiology The prevalence is unknown; about 50 cases have been reported to date. Clinical description Disease onset usually presents shortly after birth, but may present in infancy, with profound sensorineural deafness, small or absent nails, and short distal phalanges of hands and feet. About 1/4 of patients have long, finger-like thumbs. Facial features are highly variable and only a broad nasal bridge is present in 1/2 of all cases. Other facial signs, reported infrequently, include anteverted nares, long philtrum, thin upper vermilion and low set ears.