-
Naegeli-Franceschetti-Jadassohn Syndrome
OMIM
In a restudy of the original family, Franceschetti and Jadassohn (1954) documented autosomal dominant inheritance. The disorder was earlier confused with incontinentia pigmenti (IP; see 308300). ... Itin et al. (1993) reexamined the original family with NFJS 65 years after the first description (Naegeli, 1927). The pedigree included 62 members with 14 affected patients; Itin et al. (1993) examined the 10 living patients. ... NFJS and dermatopathia pigmentosa reticularis (DPR; 125595) are closely related autosomal dominant ectodermal dysplasia syndromes that clinically share complete absence of dermatoglyphics (fingerprint lines), a reticulate pattern of skin hyperpigmentation, thickening of the palms and soles (palmoplantar keratoderma), abnormal sweating, and other subtle developmental anomalies of the teeth, hair, and skin. ... History Family 1 in the study of NFJS by Lugassy et al. (2006) was the large multigenerational Swiss family in which the disorder was originally described by Naegeli in 1927 and had been followed since that time in a number of reports over a period of 80 years (Franceschetti and Jadassohn, 1954; Itin et al., 1993). INHERITANCE - Autosomal dominant HEAD & NECK Teeth - Premature tooth loss - Carious teeth - Yellow discoloration SKIN, NAILS, & HAIR Skin - Reticulate hyperpigmentation (periocular, perioral, chest, neck, abdomen) - Hypohidrosis - Absent fingerprints - Palmoplantar keratoderma - Multiple, small punctate keratoses (palms and soles) Nails - Brittle nails - Congenital malalignment of great toenails MISCELLANEOUS - Heat intolerance - Onset of hyperpigmentation in early childhood (3 months-6 years) that fades after puberty MOLECULAR BASIS - Caused by mutation in the keratin-14 gene (KRT14, 148066.0015 ) ▲ Close

-
Dupuytren Contracture
OMIM
Clinical Features Manson (1931) described affected father and 3 sons with contractures of fingers. Autosomal dominant inheritance with incomplete penetrance was likely. Under the designation 'familial fibromatosis,' Young and Fortt (1981) described a family in which at least 5 members had Dupuytren contractures. The proband was first observed to have several small lumps on his trunk at the age of 4 months. ... Mapping In a 5-generation Swedish family in which at least 17 members had Dupuytren contracture with autosomal dominant inheritance, Hu et al. (2005) identified a 6-cM candidate disease locus, referred to here as DUPC1, between markers D16S419 and D16S3032 on chromosome 16q (maximum 2-point lod score of 3.18 at marker D16S415). ... Ophoff et al. (2011) responded that their preliminary data did not support an immunologic cause of Dupuytren disease, but that testing of hypotheses such as that of Balaji et al. (2011) would lead to a greater understanding of the disorder. INHERITANCE - Autosomal dominant SKELETAL Hands - Contractures of the fingers (especially fifth finger) MUSCLE, SOFT TISSUES - Thickening of the fascial structures of the palm and fingers - Shortening of the fascial structures of the palm and fingers - Plantar fibromatosis MISCELLANEOUS - Onset in fifth or sixth decade - Earlier onset is associated with more aggressive disease course - Male to female ratio is greater than 3:1 - Reduced penetrance - Progressive disorder - High risk of recurrence after surgery - Associated with trauma and impaired wound repair (alcoholism, diabetes, substance abuse, liver disease) ▲ Close
-
Cerebellar Ataxia, Nonprogressive, With Mental Retardation
OMIM
Description Nonprogressive cerebellar ataxia with mental retardation is an autosomal dominant neurodevelopmental disorder characterized by mildly delayed psychomotor development, early onset of cerebellar ataxia, and intellectual disability later in childhood and adult life. ... Clinical Features Thevenon et al. (2012) reported 2 unrelated families and an unrelated single patient with mild mental retardation and ataxia apparent from infancy. In the first family, 2 adult half sisters had mild mental retardation, attended schools for special needs, and worked at simple jobs. ... Inheritance The transmission pattern in 2 families with nonprogressive cerebellar ataxia with mental retardation reported by Thevenon et al. (2012) was consistent with autosomal dominant inheritance. Molecular Genetics Using array CGH, Thevenon et al. (2012) identified a heterozygous intragenic deletion in the CAMTA1 gene (611501.0001) in affected members of a large family with early-onset nonprogressive cerebellar ataxia and mild mental retardation. ... Functional studies of the variant and additional studies of patient cells were not performed. INHERITANCE - Autosomal dominant HEAD & NECK Head - Large forehead - Broad forehead Face - Long face - Pointed chin - Long philtrum Ears - Short ears - Low-set ears - Prominent ears Eyes - Strabismus (in some patients) - Palpebral edema (in 1 family) - Hypertelorism - Downslanting palpebral fissures Nose - Wide flat nose - Bulbous nose - Anteverted nostrils Mouth - Thick lower lip - Small mouth Teeth - Abnormally implanted teeth (in 1 family) ABDOMEN Gastrointestinal - Constipation - Gastroesophageal reflux MUSCLE, SOFT TISSUES - Hypotonia, neonatal NEUROLOGIC Central Nervous System - Delayed psychomotor development - Mental retardation, mild - Intellectual disability - Speech delay - Unsteady gait - Ataxia - Dysmetria, mild - Dysarthria - Poor motor coordination, fine and gross - Seizures (uncommon) - Cerebellar hypoplasia - Hippocampal atrophy (in 2 siblings) - Cortical atrophy (in 2 siblings) Behavioral Psychiatric Manifestations - Behavioral difficulties (in some patients) - Attention-deficit - Hyperactivity - Aggressive behavior MISCELLANEOUS - Dysmorphic facial features are variable - Ataxia is nonprogressive MOLECULAR BASIS - Caused by disruption of the calmodulin-binding transcription activator 1 gene (CAMTA1, 611501.0001 ) ▲ Close
-
Mitochondrial Dna Depletion Syndrome 12b (Cardiomyopathic Type), Autosomal Recessive
OMIM
Heterozygous mutation in the SLC25A4 gene causes autosomal dominant progressive external ophthalmoplegia-2 (PEOA2; 609283) and autosomal dominant MTDPS12A (617184). ... Clinical Features Bakker et al. (1993) described an 8-year-old boy with adenine nucleotide translocator deficiency in muscle who was first investigated at the age of 3.5 years because of shortness of breath and rapid fatigue. ... The clinical and biochemical features were different from those found in dominant ANT1 mutations, resembling those described in ANT1-knockout mice.

-
Hereditary Pancreatitis
Wikipedia
Hereditary pancreatitis This condition is inherited in an autosomal dominant manner Hereditary pancreatitis ( HP ) is an inflammation of the pancreas due to genetic causes. It was first described in 1952 by Comfort and Steinberg [1] but it was not until 1996 that Whitcomb et al [2] isolated the first responsible mutation in the trypsinogen gene ( PRSS1 ) on the long arm of chromosome seven ( 7q35 ). ... These mutations are rarely identified in general screens of patients with idiopathic disease [10] [11] [12] [13] and the phenotype of p.R122H and p.N29I is now well characterised [4] [5] [6] with the p.A16V mutation recently characterised for the first time. [14] There are many other rare mutations or polymorphisms of PRSS1 which remain less well understood [15] [16] and not all HP families have had the responsible genetic mutation identified. ... However, a novel mechanism has recently been identified in a p.R116C kindred. [19] Diagnosis [ edit ] Families are defined as having HP, [5] if the phenotype is consistent with highly penetrant autosomal dominant inheritance. In simple terms, this would require two or more first degree relatives (or three or more second degree relatives) to have unexplained recurrent-acute or chronic pancreatitis in two or more generations. It is an autosomal dominant disease with penetrance that is generally accepted to be ≈80%. [1] [20] Management [ edit ] Treatment of HP resemble that of chronic pancreatitis of other causes.SPINK1, PRSS1, CTRC, CFTR, PRSS2, CPA1, CASR, HP, STAT6, OR10A4, NAB2, MSR1, ELAC2, TP53, PCAP, RNASEL, GAST, CBX4, NGFR, KLK3, CD274, LGR5, NPEPPS, TNF, PIK3CA, IL10, PROS1, CTSB, CRP, PIK3CB, PSAT1, PRSS3, PLAG1, BRCA2, PIK3CG, CDKN2A, COX2, PIK3CD, VEGFA, UBB, TSC2, SERPINA1, TGFB1, PTGS2, TLR4, SYP, SRD5A2, S100A8, SOX9, S100A9, MAPK3, ABL2, CLDN1, CRISPLD2, PINX1, SPHK2, SMURF1, EHMT1, CAMKMT, APOL3, PANX3, ADIPOQ, PRSS58, MIR204, HPC3, ZGLP1, MIR4270, MTCO2P12, PGPEP1, DLL4, FOXP3, MBL3P, BHLHE22, AMACR, HEY1, CELA3B, LZTS1, NCOA2, HOXB13, DLC1, HDAC6, MAGEC1, PECAM1, HDAC9, HPCX, PGR, MUC5AC, PDCD1, DSPP, CDKN1C, CP, CPB2, CLDN7, CYP1B1, CYP17A1, EEF1B2P2, CDK2, EPHB4, ERBB2, ERCC1, F13A1, FAP, FCGR2B, CDKN1B, CDH1, GCG, BDNF, APC, BIRC2, APP, RERE, CCND1, BCL2, BGN, CD40, BRAF, CALM1, CALM2, CALM3, CCKBR, KRIT1, FOXD1, GDNF, PCNA, MLH1, MAP2, MBL2, MGMT, MGST1, CD99, MIF, MMP9, LCN2, MPO, MUC1, ABO, MUC6, NGF, NOTCH1, EPCAM, KRT19, GLP1R, HDGF, GPI, GPT, GSTM1, GSTM3, GSTT1, HDAC1, HIF1A, IRF1, HLA-DQA1, HLA-DQB1, IL1A, IL1B, IL4, CXCL8, H3P10
-
Urushiol-Induced Contact Dermatitis
Wikipedia
Since the skin reaction is an allergic one, people may develop progressively stronger reactions after repeated exposures, or have no immune response on their first exposure but show sensitivity on subsequent exposures. [ citation needed ] Approximately 80% to 90% of adults will get a rash if they are exposed to 50 micrograms of purified urushiol. ... For people who have never been exposed or are not yet allergic to urushiol, it may take 10 to 21 days for a reaction to occur the first time. Once allergic to urushiol, however, most people break out 48 to 72 hours after contact with the oil. ... This is because of the underlying histamine-independent physiology of a Type IV hypersensitivity reaction . The sleepiness that first generation oral antihistamines (i.e., diphenhydramine and other first generation H1 antagonists ) produce may help people ignore the itch during the night, but do not stop nighttime scratching and may actually decrease overall sleep quality. [17] In cases of extreme symptoms, steroids such as prednisone , triamcinolone , or dexamethasone are sometimes administered to attenuate the immune response and prevent long-term skin damage, especially if the eyes are involved. ... Retrieved October 5, 2015 . ^ Smith, Huron H., 1933, Ethnobotany of the Forest Potawatomi Indians, Bulletin of the Public Museum of the City of Milwaukee 7:1-230, page 42 ^ Motz; Bowers; Young; Kinder (2012).

-
Ige Responsiveness, Atopic
OMIM
., 1928; Schwartz, 1952) proposed dominant inheritance. Demonstration of immune response genes in man (146850) gives support to the heritability of atopy (and tends to support dominant inheritance). ... From determinations of IgE levels in 29 families, Bias et al. (1973) suggested the existence of 'an autosomal dominant gene coding for a substance which represses the biosynthesis or controls the metabolism of IgE.' ... They concluded that these data were consistent with a regulatory locus for IgE occupied by 2 alleles, RE and re, with the dominant allele suppressing persistently high levels of IgE. ... In an erratum, the authors noted that the first base of the translation start site of the eotaxin 2 genomic reference sequence had been denoted as +1, introducing some errors in the numbering of the eotaxin 2 SNPs. ... Pulmonary - Asthma - Hay fever Inheritance - Autosomal dominant Immunology - Atopic hypersensitivity Lab - IgE level control Skin - Eczema ▲ Close
-
Classic Galactosemia And Clinical Variant Galactosemia
GeneReviews
If a lactose-restricted diet is provided during the first ten days of life, the severe acute neonatal complications are usually prevented. ... Treatment of manifestations: In rare instances, cataract surgery may be needed in the first year of life. Childhood apraxia of speech and dysarthria require expert speech therapy. ... Targeted analysis for common pathogenic variants can be performed first in individuals of European or African ancestry (see Table 1). ... If a lactose-restricted diet is provided during the first ten days of life, the severe acute neonatal complications are usually prevented. ... Cataract surgery may need to be performed in the first year of life, especially in the rare individuals where failure to perform NBS has resulted in delayed diagnosis.GALT, GALE, GALK1, GAL, LGALS1, AMH, GALM, SPARC, S100A1, S100A8, S100B, BEST1, UGP2, UGT8, SEPTIN4, SDF4, LGALS7B, BRD2, OTC, PAH, CASP3, MAP1B, LGALS7, LEP, IL11, IGF1, GP2, G6PD, DMD, CYP51A1, CETN1, RN7SL263P

-
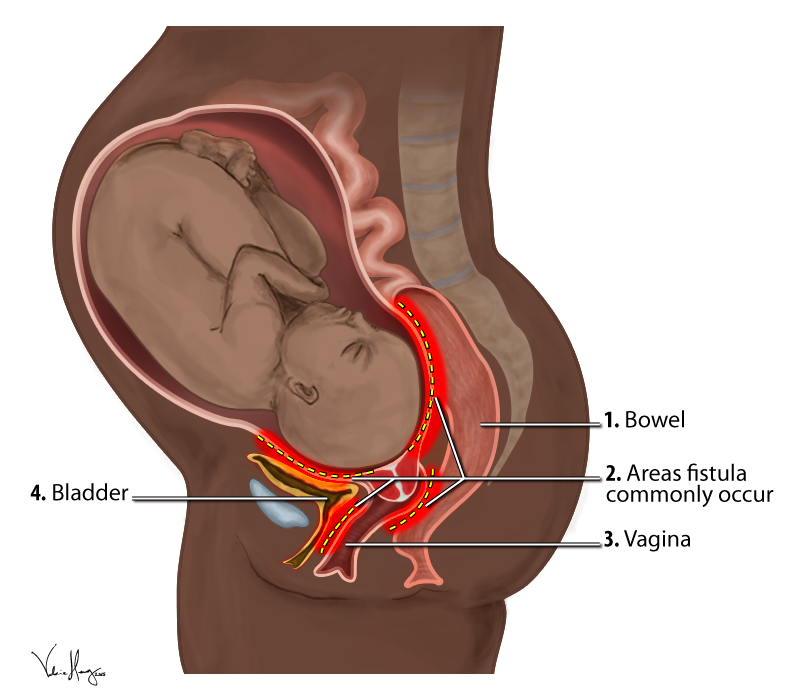
Obstetric Fistula
Wikipedia
Further, only a quarter of women who suffer a fistula in their first birth are able to have a living baby, and therefore have minuscule chances of conceiving a healthy baby later on. ... The gynecological reference in this papyrus addresses uterine prolapse, but at the end of page three, there seems to be a mention of the vesico-vaginal fistula, warning the physician against trying to cure it saying, “prescription for a woman whose urine is in an irksome place: if the urine keeps coming and she distinguishes it, she will be like this forever.” [67] This seems to be the oldest reference to vesico-vaginal fistula, one which articulates the storied history of the problem. ... This is reflected by the fact that the condition was not included as a topic at the landmark United Nations 1994 International Conference on Population and Development (ICPD). [68] The 194-page report from the ICPD does not include any reference to obstetric fistulae. ... The official international partnership formed by the Campaign to End Fistula is named the Obstetric Fistula Working Group (OFWG) and its purpose is to coordinate and collaborate global efforts to eliminate obstetric fistulae. [17] The first thing that the initiative did was to quantitatively assess the issue in countries where the prevalence is suspected to be high, including nine countries in sub-Saharan Africa. ... Retrieved April 10, 2012 . ^ Kristoff ND (2010). Half the Sky . New York: First Vintage Books. ^ Burkina Faso Ministry of Health and UNFPA.
-
Wernicke–korsakoff Syndrome
Wikipedia
For example, in France, a country that is well known for its consumption and production of wine, prevalence was only 0.4% in 1994, while Australia had a prevalence of 2.8%. [37] History [ edit ] Wernicke encephalopathy [ edit ] Carl Wernicke discovered Wernicke encephalopathy in 1881. His first diagnosis noted symptoms including paralyzed eye movements, ataxia , and mental confusion. ... ISBN 978-1-124-75696-7 . OCLC 781626781 . [ page needed ] ^ a b c d e f Sechi, GianPietro; Serra, Alessandro (2007). ... "Wernicke-Korsakoff Syndrome Following Uvulopalatopharyngoplasty for Sleep Apnea". in "Abstracts Presented at the Thirty-First Annual International Neuropsychological Society Conference, February 5–8, 2003 Honolulu, Hawaii" . ... PMID 15303623 . ^ a b c Kyoko Konishi. (2009) The Cognitive Profile of Elderly Korsakoff's Syndrome Patients. [ page needed ] ^ Caulo, M. (2005). "Functional MRI study of diencephalic amnesia in Wernicke-Korsakoff syndrome" . ... McHugh Eleanor Maguire George Armitage Miller Brenda Milner Lynn Nadel Dominic O'Brien Ben Pridmore Henry L. Roediger III Steven Rose Cosmos Rossellius Daniel Schacter Richard Shiffrin Arthur P.

-
Mathematical Anxiety
Wikipedia
Ashcraft determined that by administering a test that becomes increasingly more mathematically challenging, he noticed that even highly math-anxious individuals do well on the first portion of the test measuring performance. ... Discussion of this nomination can be found on the talk page . ( September 2013 ) ( Learn how and when to remove this template message ) Causes [ edit ] Students often develop mathematical anxiety in schools, often as a result of learning from teachers who are themselves anxious about their mathematical abilities in certain areas. ... According to John Taylor Gatto , as expounded in several lengthy books, [35] [ page needed ] modern Western schools were deliberately [ dubious – discuss ] designed during the late 19th century to create an environment which is ideal for fostering fear and anxiety, and for preventing or delaying learning. ... This is further supported by a survey of Montgomery County, Maryland students who "pointed to their parents as the primary force behind the interest in mathematics". [41] Claudia Zaslavsky [41] contends that math has two components. The first component, commonly focused on in many schools, is to calculate the answer. ... (New York: W. W. Norton & Company, 1993), page 52 ^ Murray, Margaret A. M. (2000).
-
Human Genetic Resistance To Malaria
Wikipedia
Please help improve it or discuss these issues on the talk page . ( Learn how and when to remove these template messages ) This article may be too technical for most readers to understand . ... Several inherited variants in red blood cells have become common in parts of the world where malaria is frequent as a result of selection exerted by this parasite . [3] This selection was historically important as the first documented example of disease as an agent of natural selection in humans . It was also the first example of genetically controlled innate immunity that operates early in the course of infections, preceding adaptive immunity which exerts effects after several days. ... Further details may exist on the talk page . ( April 2014 ) Sickle-cell disease was the genetic disorder to be linked to a mutation of a specific protein. ... He first delivered his hypothesis at the Eighth International Congress of Genetics held in 1948 at Stockholm on a topic "The Rate of Mutation of Human Genes". [63] He formalised in a technical paper published in 1949 in which he made a prophetic statement: "The corpuscles of the anaemic heterozygotes are smaller than normal, and more resistant to hypotonic solutions.
-
Personality Changes
Wikipedia
Please help improve it or discuss these issues on the talk page . ( Learn how and when to remove these template messages ) This article is written like a personal reflection, personal essay, or argumentative essay that states a Wikipedia editor's personal feelings or presents an original argument about a topic. ... There might be a discussion about this on the talk page . ( April 2012 ) ( Learn how and when to remove this template message ) ( Learn how and when to remove this template message ) Personality is the makeup of an individuals thinking, feeling, and behavior and is bound to change over the period of a lifetime. ... Adolescence and young adulthood have been found to be prime periods of personality changes, especially in the domains of extraversion and agreeableness. [10] It has long been believed that personality development is shaped by life experiences that intensify the propensities that led individuals to those experiences in the first place, [11] which is known as the corresponsive principle . [12] Subsequent research endeavors have integrated these findings in their methods of investigation. ... Biological transitions are stages like puberty or first childbirth. Social transitions might be changes in social roles like becoming a parent or working at a first job.PSEN1, MAPT, C9orf72, TREM2, EIF2B2, TYROBP, VCP, PLA2G6, SQSTM1, EIF2B4, EIF2B3, EIF2B5, ATXN8OS, SNCAIP, PNPLA6, VPS13A, CHMP2B, TBK1, RNF216, TMEM106B, JPH3, TBP, ADH1C, HTT, HLA-DQB1, DCTN1, TIMM8A, EIF2B1, GBA, GLUD2, GRN, ATP7B, ATXN2, LMNB1, NR4A2, PRNP, HTRA1, SNCA, LAMC2, MOG, PAEP, CSF2, BCHE
-
Coach Syndrome
Wikipedia
The condition is associated with moderate intellectual disability . [2] It falls under the category of a Joubart Syndrome -related disorder (JSRD). [3] The syndrome was first described in 1974 by Alasdair Hunter and his peers at the Montreal Children's Hospital . [4] It was not until 1989 that it was labelled COACH syndrome, by Verloes and Lambotte, at the Sart Tilman University Hospital, Belgium. [5] Contents 1 Signs and symptoms 2 Genetics 3 Diagnosis 4 Management 5 Prognosis 6 History 7 References Signs and symptoms [ edit ] An example of coloboma of the eye Signs of COACH syndrome tend to present from birth to early childhood. ... Survival depends on the severity of the symptoms, with most patients surviving infancy. [6] The likely causes of death with the progression of time are a renal and hepatic failure in later stages of life. [21] History [ edit ] The first official report of COACH syndrome was published in 1974 at the Montreal Children's Hospital , identifying two siblings, a brother and sister, presenting with all 5 components of the disorder. ... PMID 26183508 . ^ "Cerebellar Hypoplasia Information Page | National Institute of Neurological Disorders and Stroke" . www.ninds.nih.gov .
-
Multiple Drug Resistance
Wikipedia
Influenza virus has become increasingly MDR; first to amantadines, then to neuraminidase inhibitors such as oseltamivir , (2008-2009: 98.5% of Influenza A tested resistant), also more commonly in people with weak immune systems. ... Clinical Microbiology and Infection, Vol 8, Iss. 3 first published 27 July 2011 [via Wiley Online Library]. ... External links [ edit ] BURDEN of Resistance and Disease in European Nations - An EU project to estimate the financial burden of antibiotic resistance in European Hospitals European Centre of Disease Prevention and Control and (ECDC): Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance http://www.ecdc.europa.eu/en/activities/diseaseprogrammes/ARHAI/Pages/public_consultation_clinical_microbiology_infection_article.aspx State of Connecticut Department of Public Health MDRO information http://www.ct.gov/dph/cwp/view.asp?
-
Fistula
Wikipedia
Monarda fistulosa , for example, has tubular flowers. [8] The term was first used in the 14th century. [2] Contents 1 Definition 2 Classification 2.1 Location 2.2 H: Diseases of the eye, adnexa, ear, and mastoid process 2.3 I: Diseases of the circulatory system 2.4 J: Diseases of the respiratory system 2.5 K: Diseases of the digestive system 2.6 M: Diseases of the musculoskeletal system and connective tissue 2.7 N: Diseases of the urogenital system 2.8 Q: Congenital malformations, deformations and chromosomal abnormalities 2.9 T: External causes 3 Causes 4 Treatment 5 Therapeutic use 6 Epidemiology 7 Botany 8 Society and culture 9 See also 10 References 11 External links Definition [ edit ] A fistula is an abnormal connection between vessels or organs that do not usually connect. ... Monarda fistulosa , for example, has tubular flowers; [8] Eutrochium fistulosum has a tubular stem; Allium fistulosum has hollow or tubular leaves, and Acacia seyal subsp. fistula is the subspecies with hollow spines. [ citation needed ] Society and culture [ edit ] The term was first used in the 14th century. [2] See also [ edit ] Obstetric fistula Stoma (medicine) Alexis St. ... "More Funding Needed to Help Victims of Sexual Violence" ^ Emily Wax, Washington Post Foreign Service. Saturday, October 25, 2003; Page A01 "A Brutal Legacy of Congo War" External links [ edit ] Classification D MeSH : D005402 External resources MedlinePlus : 002365LAMC2, CSF2, TNF, NOD2, AKT1, CRP, ALB, IL6, IL10, IL13, TGFB1, SULT1E1, A1BG, SST, SNAI1, ZIC2, SLC22A4, SLC25A1, RASA1, RAF1, SLC22A5, TBC1D9, ZHX2, MIXL1, C20orf181, MIR340, DNAAF3, PTCRA, IL23R, ACCS, TET1, DKK1, MYH14, ACSS2, DNAI1, PLA2G15, ATP6V0A2, MAPK3, PTS, PCBD1, PLEC, F11, B2M, TSPO, CAV1, CHRM3, MAP3K8, SLC25A10, CTF1, CTLA4, ACE, DNAH5, DPEP1, EFNB2, ETS1, FAAH, ABCB1, GH1, HNF4A, HSPA1B, HSPA5, IGFBP3, IL17A, MMP2, MMP3, MMP9, NF1, NHS, PCNT, PCYT1A, CERNA3
-
Mog Antibody Disease
Wikipedia
Treatment [ edit ] Acute therapy consists of high-dose corticosteroids, IVIG, or plasma exchange, and long-term immunosuppression may be necessary in recurrent cases. [32] [33] Anti-MOG positive patients should not be treated with interferons as these may worsen the disease course similar to those with NMOSD. [26] There are also anecdotal reports against using fingolimod [34] or Alemtuzumab . [35] Research [ edit ] Animal models in experimental autoimmune encephalomyelitis, EAE , have shown that “MOG-specific EAE models (of different animal strains) display/mirror human multiple sclerosis" but EAE pathology is closer to NMO and ADEM than to the confluent demyelination observed in MS. [36] History [ edit ] Reports describing the possible involvement of anti-MOG antibodies in multiple sclerosis and other demyelinating conditions first appeared in the literature in the late 1980s, but evidence to support their role in demyelinating disease was always weak and inconsistent. [37] The possibility of an anti-MOG MS-subtype was considered around 2000. [38] The turning point was in 2011, when Mader et al. developed a cell-based assay using HEK 293 cells which increased the detection rate of these antibodies in the serum. [39] Reports about prevalence of anti-MOG in selected Multiple Sclerosis cases began to appear in 2016 [40] References [ edit ] ^ Tajfirouz, Deena A.; Bhatti, M. ... Cross-reactivity between myelin oligodendrocyte glycoprotein and human endogenous retrovirus W protein: nanotechnological evidence for the potential trigger of multiple sclerosis, Micron Volume 120, May 2019, Pages 66-73, doi: https://doi.org/10.1016/j.micron.2019.02.005 ^ Spadaro Melania; et al. (2015). ... Leite Angela Vincent, Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis, Brain, Volume 140, Issue 3, 1 March 2017, Pages 617–627, https://doi.org/10.1093/brain/aww350 , 24 February 2017 ^ Pérez CA, Garcia-Tarodo S, Troxell R.
-
Addictive Personality
Wikipedia
In these rats, a positive correlation was found between locomotor response to novel stimuli and the amount of amphetamine self-administered during the first few days of testing. [11] Twin and adoption studies have shown genetic factors account for 50-60% of the risk for alcoholism. ... HarperCollins. pp. 59– . ISBN 978-0-06-015996-2 . [ page needed ] ^ Cox, W. Miles (1985). ... Washington DC: American Psychological Association. [ page needed ] ^ Stannard, Lia (Mar 9, 2011).
-
Ciliopathy
Wikipedia
IFT88 Novel form of congenital anosmia , reported in 2012 [18] Likely ciliopathies [ edit ] Condition OMIM Gene(s) Systems/organs affected Acrocallosal syndrome [15] 200990 KIF7 , GLI3 Acromelic frontonasal dysostosis [15] 603671 ZSWIM6 Arima syndrome [15] 243910 Biemond syndrome [15] 113400 COACH syndrome [15] 216360 TMEM67 , CC2D2A , RPGRIP1L Conorenal syndrome [19] [15] 266920 Greig cephalopolysyndactyly syndrome [15] 175700 GLI3 Hydrolethalus syndrome [15] 236680 HYLS1 Johanson–Blizzard syndrome [15] 243800 UBR1 Mohr syndrome ( oral-facial-digital syndrome type 2) [15] 252100 Neu–Laxova syndrome [15] 256520 PHGDH , PSAT1 , PSPH Opitz G/BBB syndrome [15] 300000 MID1 Pallister–Hall syndrome [15] 146510 GLI3 Papillorenal syndrome [15] 120330 PAX2 Renal–hepatic–pancreatic dysplasia [15] 208540 NPHP3 Varadi–Papp syndrome (oral-facial-digital syndrome type 6) [15] 277170 Possible ciliopathies [ edit ] Condition OMIM Gene(s) Systems/organs affected Acrofacial dysostosis [15] Acrofrontofacionasal dysostosis 2 [15] 239710 Adams–Oliver syndrome [15] 100300 ARHGAP31 , DOCK6 , RBPJ , EOGT , NOTCH1 , DLL4 Asplenia with cardiovascular anomalies (Ivemark syndrome) [15] 208530 Autosomal recessive spastic paraplegia [15] Barakat syndrome (HDR syndrome) [15] 146255 GATA3 Basal cell nevus syndrome [15] 109400 PTCH1 , PTCH2 , SUFU Branchio‐oculo‐facial syndrome [15] 113620 TFAP2A C syndrome (Opitz trigonocephaly) [15] 211750 CD96 Carpenter syndrome [15] 201000 RAB23 Cephaloskeletal dysplasia (microcephalic osteodysplastic primordial dwarfism type 1) [15] 210710 RNU4ATAC Cerebrofaciothoracic dysplasia [15] 213980 TMCO1 Cerebrofrontofacial syndrome (Baraitser–Winter syndrome) [15] 243310 ACTB Cerebrooculonasal syndrome [15] 605627 Autosomal recessive spastic ataxia of Charlevoix-Saguenay [15] 270550 SACS Chondrodysplasia punctata 2 [15] 302960 EBP Choroideremia [15] 303100 CHM Chudley–McCullough syndrome [15] 604213 GPSM2 C‐like syndrome [15] 605039 ASXL1 Coffin–Siris syndrome [15] 135900 ARID1B , SOX11 , ARID2 Cohen syndrome [15] 216550 VPS13B Craniofrontonasal dysplasia [15] 304110 EFNB1 Dysgnathia complex [15] 202650 Ectrodactyly–ectodermal dysplasia–cleft syndrome type 1 [15] 129900 Endocrine–cerebroosteodysplasia syndrome [15] 612651 ICK Focal dermal hypoplasia [15] 305600 PORCN Frontonasal dysplasia [15] 136760 ALX3 , ALX4 , ALX1 Fryns microphthalmia syndrome [15] 600776 Fryns syndrome [15] 229850 Genitopatellar syndrome [15] 606170 KAT6B Hemifacial microsomia [15] 164210 Hypothalamic hamartomas [15] 241800 Johnson neuroectodermal syndrome [15] 147770 Juvenile myoclonic epilepsy [20] 254770 Kabuki syndrome [15] 147920 KMT2D , KDM6A Kallmann syndrome [15] 308700 ANOS1 Lenz–Majewski hyperostotic dwarfism [15] 151050 PTDSS1 Lissencephaly 3 [15] 611603 TUBA1A Marden–Walker syndrome [6] [15] 248700 PIEZO2 MASA syndrome [15] 303350 L1CAM Microhydranencephaly [15] 605013 NDE1 Mowat–Wilson syndrome [15] 235730 ZEB2 NDH syndrome [15] 610199 GLIS3 Oculoauriculofrontonasal syndrome [15] 601452 Oculocerebrocutaneous syndrome [15] 164180 Oculodentodigital dysplasia [15] 164200 GJA1 Optiz–Kaveggia syndrome [15] 305450 MED12 Otopalatodigital syndrome 2 [15] 304120 FLNA Periventricular heterotopia X‐linked [15] 300049 FLNA Perlman syndrome [15] 267000 DIS3L2 Pitt–Hopkins syndrome [15] 610954 TCF4 Polycystic liver disease [6] 174050 Proteus syndrome [15] 176920 AKT1 Pseudotrisomy 13 [15] 264480 Retinal cone dystrophy 1 [15] 180020 Some forms of retinitis pigmentosa [6] [21] [15] 268000 Robinow syndrome [15] 268310 ROR2 Rubinstein–Taybi syndrome [15] 180849 CREBBP Sakoda complex [15] 610871 Schinzel–Giedion syndrome [15] 269150 SETBP1 Split-hand/foot malformation 3 [15] 246560 Spondyloepiphyseal dysplasia congenita [15] 183900 COL2A1 Thanatophoric dysplasia [15] 187600 FGFR3 Townes–Brocks syndrome [15] 107480 SALL1 , DACT1 Tuberous sclerosis [15] 191100 TSC1 , TSC2 VATER association [15] 192350 Ven den Ende–Gupta syndrome [15] 600920 SCARF2 Visceral heterotaxy [15] 606325 Walker–Warburg syndrome [15] 236670 Warburg Micro syndrome [15] 615663 RAB3GAP1 X‐linked congenital hydrocephalus [15] 307000 L1CAM X‐linked lissencephaly [15] 300067 DCX Young–Simpson syndrome [15] 603736 KAT6B History [ edit ] Although non-motile or primary cilia were first described in 1898, they were largely ignored by biologists. ... PMID 17959775 . ^ of organs The Ciliary Proteome , Ciliaproteome V3.0 - Home Page, accessed 2010-06-11. ^ Hayden EC (2008). ... External links [ edit ] Classification D MeSH : D002925 DiseasesDB : 29887 External resources Orphanet : 363250 The Ciliary Proteome Web Page at Johns Hopkins v t e Diseases of cilia Structural receptor: Polycystic kidney disease cargo: Asphyxiating thoracic dysplasia basal body : Bardet–Biedl syndrome mitotic spindle : Meckel syndrome centrosome : Joubert syndrome Signaling Nephronophthisis Other/ungrouped Alström syndrome Primary ciliary dyskinesia Senior–Løken syndrome Orofaciodigital syndrome 1 McKusick–Kaufman syndrome Autosomal recessive polycystic kidney See also: ciliary proteinsWDR19, OFD1, KIF7, CEP290, TMEM67, RPGRIP1L, BBS2, DYNC2H1, MKS1, AHI1, MKKS, ARL13B, CPLANE1, IQCB1, NEK1, NEK8, CC2D2A, RPGR, SDCCAG8, TTC21B, ZNF423, NPHP1, NPHP3, WDR35, CEP164, PKHD1, RPGRIP1, LCA5, IFT80, XPNPEP3, TULP1, EVC2, BBS5, GLIS2, CRB1, UMOD, TMEM231, TMEM216, CCDC28B, BBS9, TCTN2, INVS, B9D1, TRIM32, FAM92A, TMEM237, USH2A, BBS1, NPHP4, BBS4, GUCY2D, GDF1, PCARE, TOPORS, USH1C, NR1H4, CCDC40, DNAAF2, LRAT, B9D2, FOXH1, DNAH11, ADGRV1, ARL6, IMPDH1, CDH23, BBS10, EVC, CCDC39, WDPCP, DNAAF3, CRELD1, CFTR, CRX, NKX2-5, TMEM138, RD3, PCDH15, DNAH5, TCTN1, WHRN, ATXN10, AIPL1, DNAH8, ZIC3, RDH12, VHL, BBS12, SCNN1A, CLRN1, SPATA7, MYO7A, CEP41, IFT43, PKD2, SCNN1G, LRRC56, TTC8, DNAAF1, USH1G, NODAL, ACVR2B, SCNN1B, RPE65, LEFTY2, BBS7, KCNJ13, HYLS1, TSC2, TSC1, SCLT1, WDR11, NEK4, IFT140, INPP5E, KIAA0586, CEP120, IFT122, IFT52, ALMS1, MAK, TMEM107, IFT172, PRKD1, DCDC2, FGFR1OP, CEP104, STK11, WDR60, TCTN3, C2CD3, CILK1, POC1B, IFT27, TGFB1, RHO, ARMC9, CFAP410, CRB2, FOXJ1, CENPF, MGS, USP35, ARL13A, NPHP3-ACAD11, TTC26, MCIDAS, MICAL3, DNAAF4, PIFO, FOPNL, NEK9, IFT20, UCN2, CEP19, TAPT1, USP38, CPLANE2, TBC1D32, FAM161A, TEKT1, SLC41A1, BICC1, CSPP1, TXNDC15, FAM149B1, ACTB, NINL, ADAMTS9, PPT1, MCM2, RAB8A, NME3, NOTCH2, ORC1, PAFAH1B1, PAM, PCM1, PCNT, PCP4, PDE6D, PIK3CA, PIK3CB, PIK3CD, PIK3CG, KPNA3, KIF11, JBS, CTNNB1, AGXT, ATD, ATR, CCND1, BUB1B, CETN2, DAP, IGF1, E2F4, FGF8, FGFR3, GAS8, GLI1, GLI2, PKD1, RAF1, CEP72, RFX1, WDR5, ACVR1, RCOR1, ARL2BP, POC1A, CHTOP, TRAF3IP1, SGSM3, IFT81, PIK3R4, HSPB11, SUFU, RAB23, IFT57, CEP55, B4GAT1, B3GNT2, KIF14, IFT88, RFX3, RNASE3, CCL2, HNF1A, TNF, EZR, TRRAP, CEP350, IKBKG, CDK10, TNFSF14, UNC119, USP8, KIAA0753, LCA10

-
Androgen Insensitivity Syndrome
OMIM
They displayed some pubic hair. The first type included a patient with the 'hairless female' phenotype, also pictured by McKusick (1964). ... In normal males, testosterone and LH rise during the first few months of life, and this physiological surge is commonly used to evaluate the gonadotropic axis at this age. ... Bouvattier et al. (2002) sequentially measured plasma testosterone, LH, and FSH during the first 3 months of life in 15 neonates with AIS and AR mutations. ... They determined participant knowledge of CAIS as well as opinion of medical and surgical treatment. As a whole, secondary sexual development of these women was satisfactory, as judged by both participants and physicians. ... Morris (1953), in a classic paper, first used the term testicular feminization.AR, FKBP4, SMS, PLAT, RSS, MTNR1B, LBX1, ESR1, LEP, MATN1, IL6, EGFR, ESR2, ADGRG6, PAICS, GART, MTHFR, C20orf181, IGF1, CALM1, F5, TPH1, MT2A, AMH, MTNR1A, MMP3, TSPAN33, TP53, TMPRSS6, BNC2, TNFRSF11B, SIRT1, NTF3, SLC39A8, NCOA2, FBN1, FBN2, LEPR, PYCARD, PAX1, PDXP, GPER1, GPX3, SERPINE1, TIMP2, SHBG, BRD2, TGFB1, SULT1E1, MIR494, VDR, SRY, STS, VEGFA, BDNF, MIR15A, EOS, TP63, ADIPOQ, CST3, IS1, PITX1, SOCS3, POC5, PON1, CTNNB1, PROM1, KAT7, GPR50, PDAP1, SIRT5, TUSC2, PAPOLA, NDRG1, TXN, ADAMTS13, MSC, MRPS30, AKR1C3, CHL1, BEST1, VWF, MYBBP1A, USP8, SMUG1, UXT, TNFSF11, ASAP2, GDF15, AANAT, TMEFF2, MIR145, DPP9, NLRP3, OCIAD2, HJV, NEAT1, C17orf67, SPATA21, MIRLET7I, MIR126, MIR130A, MIR134, MIR183, HECTD1, MIR185, MIR191, MIR192, MIR222, MIR93, PALM2AKAP2, MT1IP, MIR675, CDKN2B-AS1, MIR4300, OCLN, ADGRG7, IL17RC, DOT1L, SPRY4, SETBP1, CNTNAP2, PELP1, CD274, TBX21, ASAP1, ADIPOR1, APH1A, CLEC1B, TNF, MTPAP, LAPTM4B, SOX6, MIB1, PCDH10, MIER1, MID1IP1, SOX17, NUCKS1, AHNAK, IRX1, FUZ, VANGL1, HSD17B7, PSMD4, TIMP1, DLST, CLTC, COL4A2, COL11A1, COL11A2, COMP, MAP3K8, CREBBP, CRP, CYP2C19, DBP, DMD, CHI3L1, DPP4, DUSP2, EPO, F2, F3, FGFR3, FGR, FN1, NR5A1, GAD1, CLU, CDKN2A, THRSP, ASL, ACP3, ACTB, ADRA1D, ALB, APC, APOD, APOE, ARF6, ARG1, ARSF, ATP2A2, CDH13, ATP2B4, BGLAP, BMP4, BTF3P11, CALCA, CALM2, CALM3, CASP3, RUNX2, CD38, GC, GHSR, MSH6, ABO, MT1L, MT1X, NRGN, REG3A, PAX3, PBX1, PLG, PMCH, MAPK7, PSD, RARB, HDAC2, PRPH2, S100A12, SFPQ, SRSF1, ITSN1, SLC4A1, SOX9, SRD5A2, STAT4, TGM2, MT1M, MT1JP, MT1H, MT1G, HGF, HOXA10, HSPG2, IGFBP7, IL1A, IL1B, IL5, IL10, ITGA2B, KLK1, KRAS, LCN2, LGALS1, LGALS3, LRPAP1, KITLG, MKI67, MT1A, MT1B, MT1E, MT1F, H3P10









