In Taiwan , about 30% of newborns with elevated levels of phenylalanine have a deficiency of THB. [8] Subclinical deficiency can be found in individuals with poor diet (including low intake of folate or vitamin C ) or genetic mutations in the MTHFR genes, which are involved in BH4 synthesis and recycling. [ citation needed ] See also [ edit ] Phenylketonuria (PKU) Tetrahydrobiopterin (THB, BH 4 ) References [ edit ] ^ a b Ponzone A, Spada M, Ferraris S, Dianzani I, de Sanctis L (2004). ... PMID 12003346 . ^ Schaub J, Däumling S, Curtius HC, Niederwieser A, Bartholomé K, Viscontini M, Schircks B, Bieri JH (1978). "Tetrahydrobiopterin therapy of atypical phenylketonuria due to defective dihydrobiopterin biosynthesis" .
Dehydratase deficiency or pterin-4 alpha-carbinolamine dehydratase (PCD) is considered a transient and benign form of hyperphenylalaninemia due to tetrahydrobiopterin deficiency (see this term), characterized by muscular hypotonia, irritability (detected by EEG), slow acquisition of psychomotor skills, age-dependent movement disorders, including dystonia and an accompanying excretion of 7-substituted pterins. Neurological developement is normal with dietary control of blood phenyalanine. PCD is inherited in an autosomal recessive manner.
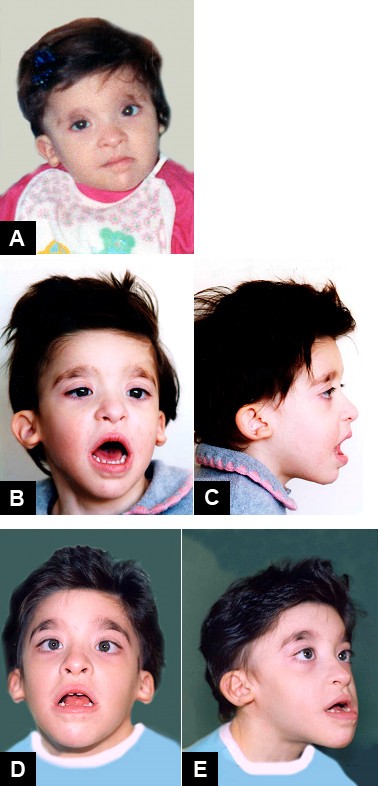
Clinical description The typical characteristic facies of MWS includes a high forehead, frontal bossing, large eyebrows that are medially flaring and sparse in the middle part, hypertelorism, deepset but large eyes, large and uplifted ear lobes with a central depression, saddle nose with prominent rounded nasal tip, prominent columella, open mouth with M-shaped upper lip, and a prominent but narrow and triangular pointed chin.
Mowat-Wilson syndrome is a genetic condition that affects many parts of the body. Major signs of this disorder frequently include distinctive facial features, intellectual disability, delayed development, an intestinal disorder called Hirschsprung disease, and other birth defects. Children with Mowat-Wilson syndrome have a square-shaped face with deep-set, widely spaced eyes . They also have a broad nasal bridge with a rounded nasal tip; a prominent and pointed chin ; large, flaring eyebrows; and uplifted earlobes with a dimple in the middle. These facial features become more distinctive with age, and adults with Mowat-Wilson syndrome have an elongated face with heavy eyebrows and a pronounced chin and jaw.
Summary Clinical characteristics. Mowat-Wilson syndrome (MWS) is characterized by distinctive facial features (widely spaced eyes, broad eyebrows with a medial flare, low-hanging columella, prominent or pointed chin, open-mouth expression, and uplifted earlobes with a central depression), congenital heart defects with predilection for abnormalities of the pulmonary arteries and/or valves, Hirschsprung disease or chronic constipation, genitourinary anomalies (particularly hypospadias in males), and hypogenesis or agenesis of the corpus callosum. Most affected individuals have moderate-to-severe intellectual disability. Speech is typically limited to a few words or is absent, with relative preservation of receptive language skills. Growth restriction with microcephaly and seizure disorder are also common. Most affected people have a happy demeanor and a wide-based gait that can sometimes be confused with Angelman syndrome.
Mowat-Wilson syndrome (MWS) is a rare genetic disorder that affects many systems of the body. Some of the main features include intellectual disability, distinctive facial features, delayed development, and Hirschsprung disease . Other features may include microcephaly, structural brain abnormalities, epilepsy, short stature, and defects of the heart, urinary tract, or genitalia. MWS is caused by a mutation in the ZEB2 gene. It typically occurs for the first time in a person with MWS and is not inherited from a parent. Vary rarely, more than one child in a family will have MWS. Treatment depends on the symptoms present and focuses on the specific needs of each person.
A number sign (#) is used with this entry because Mowat-Wilson syndrome (MOWS) is caused by de novo heterozygous mutation in the ZEB2 gene (605802) on chromosome 2q22. Description Mowat-Wilson syndrome is an autosomal dominant complex developmental disorder; individuals with functional null mutations present with mental retardation, delayed motor development, epilepsy, and a wide spectrum of clinically heterogeneous features suggestive of neurocristopathies at the cephalic, cardiac, and vagal levels. Mowat-Wilson syndrome has many clinical features in common with Goldberg-Shprintzen syndrome (609460) but the 2 disorders are genetically distinct (Mowat et al., 2003). Goldberg-Shprintzen syndrome is caused by mutation in the KIAA1279 gene (609367) located on 10q. Clinical Features Mowat et al. (1998) described 6 unrelated children with a distinctive facial phenotype in association with mental retardation, microcephaly, and short stature.
Molecular Genetics Using Sanger sequencing of the mitochondrial genome in a patient with mitochondrial myopathy, lactic acidosis, and sideroblastic anemia, Burrage et al. (2014) detected an apparently homoplasmic novel variant in the MTATP6 gene (m.8969G-A, S148N; 516060.0012). Next-generation sequencing confirmed the presence of this variant at 96% and 88% heteroplasmy in the proband's blood and muscle specimens, respectively, and did not detect the variant in his mother's blood sample.
A number sign (#) is used with this entry because of evidence that myopathy, lactic acidosis, and sideroblastic anemia-1 (MLASA1) is caused by homozygous mutation in the PUS1 gene (608109) on chromosome 12q24. Description Myopathy, lactic acidosis, and sideroblastic anemia (MLASA) is a rare autosomal recessive oxidative phosphorylation disorder specific to skeletal muscle and bone marrow (Bykhovskaya et al., 2004). Genetic Heterogeneity of Myopathy, Lactic Acidosis, and Sideroblastic Anemia MLASA2 (613561) is caused by mutation in the YARS2 gene (610957) on chromosome 12p11. MLASA3 (500011) is caused by heteroplasmic mutation in the mitochondrially-encoded MTATP6 gene (516060). Clinical Features Rawles and Weller (1974) reported 2 brothers with myopathy, lactic acidosis, and sideroblastic anemia with ringed sideroblasts.
A number sign (#) is used with this entry because of evidence that myopathy, lactic acidosis, and sideroblastic anemia-2 (MLASA2) is caused by homozygous mutation in the YARS2 gene (610957) on chromosome 12p11. Description Myopathy, lactic acidosis, and sideroblastic anemia-2 is an autosomal recessive disorder of the mitochondrial respiratory chain. The disorder shows marked phenotypic variability: some patients have a severe multisystem disorder from infancy, including cardiomyopathy and respiratory insufficiency resulting in early death, whereas others present in the second or third decade of life with sideroblastic anemia and mild muscle weakness (summary by Riley et al., 2013). For a discussion of genetic heterogeneity of MLASA, see MLASA1 (600462). Clinical Features Sasarman et al. (2002) reported a 34-year-old man of Lebanese descent (patient E) with a lifelong history of muscle weakness, exercise intolerance, occasional muscle cramping and stiffness after running, and small muscles.
Mitochondrial myopathy and sideroblastic anemia belongs to the heterogeneous family of metabolic myopathies. It is characterised by progressive exercise intolerance manifesting in childhood, onset of sideroblastic anaemia around adolescence, lactic acidaemia, and mitochondrial myopathy. Epidemiology Less than 10 cases have been described so far. Etiology A 656C-->T mutation in the nuclear pseudouridine synthase 1 gene ( PUS1 ), localised to 12q24.33, has recently been identified in some patients. Deficient pseudouridylation of mitochondrial tRNAs may be responsible for the oxidative phosphorylation disorder. Diagnostic methods Muscle biopsy demonstrates low activity of complexes 1 and 4 of the respiratory chain and paracrystalline inclusions can be revealed in most mitochondria by electron microscopy.
History [ edit ] This condition was first described in 2003. [3] References [ edit ] ^ Lessel D, Vaz, B, Halder S, Lockhart PJ, Marinovic-Terzic I, Lopez-Mosqueda J, Philipp M, Sim JCH, Smith KR, Oehler J, Cabrera, E, Freire R, et al (2014) Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features.
A rare inherited cancer-predisposing syndrome characterized by early-onset hepatocellular carcinoma, genomic instability, and progeroid features, such as short stature, low body weight, muscular atrophy, lipodystrophy, bilateral cataracts, and premature hair graying. Dysmorphic craniofacial features include triangular face, small, deep-set eyes, and micrognathia. Kyphoscoliosis, sloping shoulders, mild pectus excavatum, bilateral contractures of the elbows and fingers, bilateral clinodactyly, and pes planus have also been reported.
In addition, patient cells were hypersensitive to replication-related genotoxic agents but not to ionizing radiation, consistent with a severe G2/M-checkpoint defect. Exclusion Studies Ruijs et al. (2003) sequenced the WRN gene in a Moroccan boy with progeroid features who died of hepatocellular carcinoma at age 17 years, but detected no mutation in the coding region.
A number sign (#) is used with this entry because of evidence that some cases of neurofibromatosis-Noonan syndrome are caused by heterozygous mutation in the neurofibromin gene (NF1; 613113) on chromosome 17q11. Allelic disorders include classic neurofibromatosis type I (162200) and Watson syndrome (193520). Clinical Features Allanson et al. (1985) reported 4 unrelated patients with neurofibromatosis who had manifestations of Noonan syndrome (163950), including short stature, ptosis, midface hypoplasia, webbed neck, learning disabilities, and muscle weakness. Family history was negative in each case. Average paternal and maternal ages were 37 and 28 years, respectively, at the birth of the patients, suggesting new dominant mutation. The chromosomes, including prometaphase preparations in 3 of the 4, were normal.
A number sign (#) is used with this entry because of evidence that Watson syndrome (WTSN) is caused by heterozygous mutation in the NF1 gene (613113) on chromosome 17q11. Description Watson syndrome is an autosomal dominant disorder characterized by pulmonic stenosis, cafe-au-lait spots, decreased intellectual ability (Watson, 1967), and short stature (Partington et al., 1985). Most affected individuals have relative macrocephaly and Lisch nodules and about one-third of those affected have neurofibroma (Allanson et al., 1991). Clinical Features Watson (1967) described 15 persons from 2 generations of each of 3 families with pulmonic stenosis (8/15), cafe-au-lait spots (15/15) and low normal or dull intelligence (12/15). There were 8 males and 7 females; male-to-male transmission was noted.
Neurofibromatosis-Noonan syndrome (NFNS) is a RASopathy and a variant of neurofibromatosis type 1 (NF1) characterized by the combination of features of NF1, such as café-au-lait spots, iris Lisch nodules, axillary and inguinal freckling, optic nerve glioma and multiple neurofibromas, and Noonan syndrome (NS), such as short stature, typical facial features (hypertelorism, ptosis, downslanting palpebral fissures, low-set posteriorly rotated ears with a thickened helix, and a broad forehead), congenital heart defects and unusual pectus deformity. As these three entities have significant phenotypic overlap, molecular genetic testing is often necessary for a correct diagnosis (such as when café-au-lait spots are present in patients diagnosed with NS).
Mendelian susceptibility to mycobacterial diseases (MSMD) due to partial STAT1 (signal transducer and activator of transcription 1) deficiency is a genetic variant of MSMD (see this term) characterized by a partial defect in the interferon (IFN)-gamma pathway, leading to mild mycobacterial infections. Epidemiology The prevalence is unknown. In 2001, two patients from unrelated kindreds were described and they suffered only mild MSMD. Since 2001, up to six other kindreds have been described to have a partial dominant STAT1 deficiency associated with MSMD. Clinical description First infections occur after the age of 3, most commonly with weakly virulent Mycobacterium bovis BCG and Mycobacterium avium complex or with the more virulent Mycobacterium tuberculosis . Clinical penetrance is incomplete and some patients are asymptomatic while others have very mild clinical manifestations.
Antilymphocyte globulin (ALG) or antithymocyte globulin (ATG) is mixed with cyclosporine to promote cell growth. [7] See also [ edit ] Polycythemia , the opposite of anemia References [ edit ] ^ “Figure 2f from: Irimia R, Gottschling M (2016) Taxonomic Revision of Rochefortia Sw.
Umbrella term for deadly disease, especially of livestock Not to be confused with Morraine , a geological feature. Murrain / ˈ m ɜːr ɪ n / is an antiquated term for various infectious diseases affecting cattle and sheep . [1] It literally means "death" and was used in medieval times to represent just that. [2] Murrain did not refer to a specific disease, but was an umbrella term for what are now recognized as a number of different diseases, including rinderpest , erysipelas , foot-and-mouth disease , anthrax , and streptococcus infections.
Retrieved 2016-03-31 . ^ a b Shah, AA; Shaikh, H; Karim, M (February 1994). "Amoebic brain abscess: a rare but serious complication of Entamoeba histolytica infection" .
The injury is caused by severe external rotation of the ankle. [1] The ankle remains externally rotated after the injury, making interpretation of X-rays difficult which can lead to misdiagnosis and incorrect treatment. [2] The injury is most commonly treated by open reduction internal fixation as closed reduction is made difficult by the entrapment of the fibula behind the tibia. [1] The entrapment of an intact fibula behind the tibia was described by Ashhurst and Bromer in 1922, who attributed the description of the mechanism of injury to Huguier's 1848 publication. [3] The injury involving fibular fracture with posterior dislocation was described by David M. Bosworth in 1947. [4] References [ edit ] ^ a b Perry, CR; Rice S; Rao A; Burdge R.
It usually affects craniofacial bones, mandible most frequently, long bones (metaphyseal femur, tibia, humerus). [1] Although it does not tend to metastatize , it has a high local recurrence and infiltrative growth. [2] Treatment consists in wide local excision to prevent otherwise frequent recurrences. [3] The role of radiotherapy and chemotherapy in this tumor still is unclear. [4] Some cases have been described, in which an osteosarcoma has arisen from a desmoplastic fibroma . [5] A famous occurrence of this particular form of the disease involved Italo-Australian Riccardo Torresan in 2011, with 18 cm of femur needing to be removed with the now widely recognized method of "aggressive curettage" being employed. [6] See also [ edit ] Desmoplasia References [ edit ] ^ "Desmoplastic Firboma" . ^ Schneider, M.; Zimmermann, A. C.; Depprich, R.
Urban et al. (1979) described 2 brothers, aged 19 years and 9 months and 16 years and 8 months, with a previously undescribed condition characterized by genital anomalies, mental retardation, obesity, contractures of fingers, and osteoporosis. Pagnan and Gollop (1988) described a 12-year-old boy with a similar phenotype. The parents were nonconsanguineous. Both the fingers and the toes showed camptodactyly. In addition to osteopenia, the bones of the hands and feet showed tubulation defects and large epiphyses. GU - Genital anomalies Radiology - Osteoporosis - Tubulation defects and large epiphyses in hands and feet Neuro - Mental retardation Inheritance - Autosomal recessive Limbs - Camptodactyly Growth - Obesity - Short stature ▲ Close
Prader-Willi habitus, osteopenia, and camptodactyly syndrome is characterized by intellectual disability, short stature, obesity, genital abnormalities, and hand and/or toe contractures. It has only been described in two brothers and in one isolated case in a different family. Other symptoms included unusual face, deformity of the spinal column, osteoporosis and a history of frequent fractures. It is similar to Prader-Willi syndrome , but the authors concluded that it is a different condition. The cause was unknown in the reported cases.
This syndrome is characterized by intellectual deficit, short stature, obesity, genital abnormalities, and hand and/or toe contractures. It has been described in two brothers and in one isolated case. The patients also present with generalized osteoporosis and a history of frequent fractures. This syndrome is similar to Prader-Willi syndrome, but the hand contractures and osteoporosis, together with the lack of hypotonia, indicate this is a different entity.