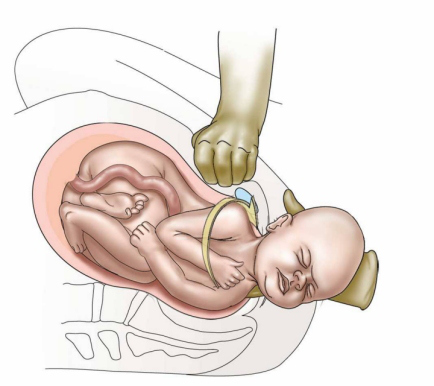
The ventral roots (motor pathway) are most prone to injury. [ citation needed ] The cause of injury to the baby is debated, [ citation needed ] but a probable mechanism is manual stretching of the nerves, which in itself can cause injury. [ citation needed ] Excess tension may physically tear the nerve roots out from the neonatal spinal column, resulting in total dysfunction. [ citation needed ] Possible complications include: Neonatal complications: Klumpke paralysis Erb's palsy Hypoxia Death Cerebral palsy Maternal complications: [5] Postpartum bleeding (11%) Perineal lacerations that extend into the anal sphincter Pubic symphysis separation Neuropathy of lateral femoral cutaneous nerve Uterine rupture Risk factors [ edit ] About 16% of deliveries where shoulder dystocia occurs have conventional risk factors. [ citation needed ] These include diabetes , [6] fetal macrosomia , and maternal obesity . [7] [8] Risk factors: [9] Age >35 Short in stature Small or abnormal pelvis More than 42 weeks gestation Estimated fetal weight >4,500 g Maternal diabetes (2–4 fold increase in risk) Factors which increase the risk/are warning signs: [ citation needed ] Need for oxytocics Prolonged first or second stage of labour Turtle sign (head bobbing in the second stage) Failure to restitute No shoulder rotation or descent Instrumental delivery For women with a previous shoulder dystocia, the risk of recurrence is at least 10%. [5] Management [ edit ] The steps to treating a shoulder dystocia are outlined by the mnemonic ALARMER: [10] A sk for help. [2] This involves asking for the help of an obstetrician, anesthesia, and for pediatrics for subsequent resuscitation of the infant that may be needed if the methods below fail; L eg hyperflexion and abduction at the hips ( McRoberts maneuver ); [2] A nterior shoulder disimpaction (suprapubic pressure); [10] R otation of the shoulder ( Rubin maneuver ); [10] M anual delivery of posterior arm; [10] E pisiotomy; [10] R oll over on all fours. [10] Typically the procedures are performed in the order listed and the sequence ends whenever a technique is successful. [10] Intentional fracturing of the clavicle is another possibility at non-operative vaginal delivery prior to Zavanelli's maneuver or symphysiotomy , both of which are considered extraordinary treatment measures. ... The Global Library of Women's Medicine . doi : 10.3843/GLOWM.10137 . ^ a b c d e f g h i j k l m n Dahlke, JD; Bhalwal, A; Chauhan, SP (June 2017). ... Retrieved 3 October 2018 . ^ Gilstrop, M; Hoffman, MK (December 2016). "An Update on the Acute Management of Shoulder Dystocia".
.; Crozier, J.; Boa, E.; Rutherford, M.; Smith J.J. (2004). "First report of Xanthomonas campestris pv. musacearum on banana in Uganda" . ... Phytopathology . 58 : 111–112. ^ Mwangi, M.; Bandyopadhyay, R.; Ragama,P.; Tushemereirwe, R.K. (2007). ... ISSN 0031-949X . PMID 31922946 . ^ Biruma, M.; et al. (2007). "Banana Xanthomonas wilt: a review of the disease, management strategies and future research directions".
Epilepsy Foundation . Retrieved 31 August 2016 . ^ Amit M. Shelat (27 February 2016). "Partial (focal) seizure" . ... Lescher 2000, p. 1749. ^ a b c Murro, Anthony M. 2006. ^ Engelsen, B A., C Tzoulis, B Karlsen, A Lillebø, L M 2008. ^ Trescher, William H., and Ronald P.
Meningococcal group C conjugate vaccine are also used in some cases. [22] In primary biliary cirrhosis ursodeoxycholic acid helps the bloodstream remove bile which may increase survival in some affected individuals. [23] See also [ edit ] Hepatosplenomegaly Liver function tests References [ edit ] ^ a b c d e f g h i j k l m n o p q r s "Hepatomegaly. Read about Hepatomegaly (enlarged liver) | Patient" . ... Elsevier Health Sciences. p. 991. ISBN 9780323322850 . ^ Meacock, L M; Sellars, M E; Sidhu, P S (2010-07-01).
Overview An enlarged liver is one that's bigger than normal. The medical term is hepatomegaly (hep-uh-toe-MEG-uh-le). Rather than a disease, an enlarged liver is a sign of an underlying problem, such as liver disease, congestive heart failure or cancer. Treatment involves identifying and controlling the cause of the condition. Enlarged liver An enlarged liver can have many possible causes. Symptoms An enlarged liver might not cause symptoms. When enlarged liver results from liver disease, it might be accompanied by: Abdominal pain Fatigue Nausea and vomiting Yellowing of the skin and the whites of the eyes (jaundice) When to see a doctor Make an appointment with your doctor if you have symptoms that worry you.
PMC 2748736 . PMID 19210057 . ^ Heilig M, Egli M, Crabbe JC, Becker HC (April 2010). ... PMID 15963001 . ^ Mirijello A, D'Angelo C, Ferrulli A, Vassallo G, Antonelli M, Caputo F, Leggio L, Gasbarrini A, Addolorato G (March 2015).
. ^ Klippel–Trenaunay syndrome: Spectrum and management ^ Tian XL, Kadaba R, You SA, Liu M, Timur AA, Yang L, Chen Q, Szafranski P, Rao S, Wu L, Housman DE, DiCorleto PE, Driscoll DJ, Borrow J, Wang Q (2004). ... PMC 1476812 . PMID 14327016 . ^ Cohen, M. Michael (2000). "Klippel–Trenaunay syndrome". ... Phlebology . 20 (2): 63–81. doi : 10.1258/0268355054069188 . ^ synd/1812 at Who Named It? ^ Klippel M, Trénaunay P (1900). "Du naevus variqueux ostéohypertrophique".
A number sign (#) is used with this entry because of evidence that capillary malformation-arteriovenous malformation-1 (CMAVM1) is caused by heterozygous mutation in the RASA1 gene (139150) on chromosome 5q14. Description Capillary malformation-arteriovenous malformation-1 is an autosomal dominant disorder characterized by atypical capillary malformations (CMs), often in association with fast-flow vascular malformations, including arteriovenous malformations (AVMs) and arteriovenous fistulas (AVFs), and Parkes Weber syndrome (PKWS). The CMs are usually multifocal and are surrounded by a pale halo with a central red dot; they increase in number with age. The AVMs generally occur in the brain or on the face or extremities. Intracranial AVMs include vein of Galen aneurysmal malformations (VGAMs). Parkes Weber syndrome is a specific type of CMAVM that presents with limb overgrowth, more commonly affecting one of the lower extremities (Eerola et al., 2003; Revencu et al., 2013; Johnson and Navarro, 2017).
Clinical Features The features of Klippel-Trenaunay-Weber syndrome are large cutaneous hemangiomata with hypertrophy of the related bones and soft tissues. The disorder clinically resembles Sturge-Weber syndrome (185300), and indeed the 2 have been associated in some cases (Harper, 1971). Lindenauer (1965) described a brother and sister with Klippel-Trenaunay syndrome. Both patients had varicosity, hypertrophy, and hemangioma, but no arteriovenous fistula. Lindenauer (1965) suggested that patients who also have arteriovenous fistula have a different disorder that might be called Parkes Weber syndrome, since Weber (1907) described cases of this type as well as cases seemingly identical to those of Klippel and Trenaunay (1900).
Klippel-Trenaunay syndrome (KTS) is a syndrome that affects the development of blood vessels, soft tissues, and bones. This syndrome has three characteristic features: a red birthmark called a port-wine stain , overgrowth of soft tissues and bones, and vein malformations such as varicose veins or malformations of deep veins in the limbs. The overgrowth of bones and soft tissues usually begins in infancy and is most often only affects one leg. However, it can also affect the arms or sometimes the upper body area (torso). The overgrowth can cause pain, a feeling of heaviness, and make the affected leg (or arm) hard to move.
A congenital vascular bone syndrome (CVBS) characterized by the presence of a vascular malformation in a limb, mainly of the arteriovenous type, which results in overgrowth of the affected limb. Epidemiology Prevalence is unknown but around 1,000 cases have been reported in the literature so far. Clinical description The affected limb may show overgrowth in comparison with the contralateral limb and the extent of this limb length discrepancy (LLD) may vary from a slight difference to 10 cm or more. The growth effect may be manifested in only one bone (mainly the femur or tibia) or, in some cases, affect the whole limb. The LLD may become apparent during infancy, childhood or adolescence and is clearly visible by comparison of the level of the gluteal and posterior knee folds.
Overview Klippel-Trenaunay (klih-PEL tray-no-NAY) syndrome ― also called KTS ― is a rare disorder found at birth (congenital) involving problems in the development of certain blood vessels, soft tissues (such as skin and muscles), bones and sometimes the lymphatic system. The main features include a red birthmark (port-wine stain), ranging in color from pink to reddish-purple, atypical vein or lymphatic development (malformations), and overgrowth of tissues and bones. These findings most often affect one leg but may occur in an arm or elsewhere. Although there is no cure for KTS , treatment goals are to improve symptoms and prevent complications. Symptoms People who have KTS may have the following features, which can range from mild to more extensive: Port-wine stain.
Klippel-Trenaunay syndrome is a condition that affects the development of blood vessels, soft tissues (such as skin and muscles), and bones. The disorder has three characteristic features: a red birthmark called a port-wine stain, abnormal overgrowth of soft tissues and bones, and vein malformations. Most people with Klippel-Trenaunay syndrome are born with a port-wine stain. This type of birthmark is caused by swelling of small blood vessels near the surface of the skin. Port-wine stains are typically flat and can vary from pale pink to deep maroon in color.
Origin of Renfield's syndrome [ edit ] The syndrome is named after R. M. Renfield , Dracula 's human zoophagous follower in the 1897 novel by Bram Stoker . ... Television and internet video [ edit ] In an NBC pre-Halloween special hosted by actor Peter Graves entitled "The Unexplained: Witches, Werewolves and Vampires" that aired on 23 October 1994 (and is available on YouTube, with the 34 m 11 s mark beginning the segment), pages from Noll's book were shown on camera as Canadian psychologist Leonard George summarized Renfield's syndrome for a wide television audience. [12] [13] Characters suffering from Renfield's Syndrome have appeared on television. ... Piercing the Darkness: Undercover with Vampires in America Today . New York: HarperTorch. ^ White, M; Omar, H (2010). "Vampirism, vampire cults and the teenager of today".
.; Domchek, S. ; Anderson, W. F.; Bartlett, J. M. S.; Gelmon, K.; Nahleh, Z.; Bergh, J.; Cutuli, B.; Pruneri, G.; McCaskill-Stevens, W.; Gralow, J.; Hortobagyi, G.; Cardoso, F.; et al. (2010). ... Retrieved 2010-05-15 . ^ a b c d e f g Gómez-Raposo, C.; Zambrana Tévar, F.; Sereno Moyano, M.; Casado, Enrique; et al. (2010). ... PMID 19427229 . . ^ Kalyani, R.; Days, S.; Bindra Singh, M. S.; Kumar, H. (2010). "Cancer profile in Kolar: A ten years study".
See also [ edit ] Medicine portal Our Curse , an Oscar-nominated 2013 short documentary film about a child with Ondine's curse References [ edit ] ^ Jazeela Fayyaz, DO (2017-12-05). Zab Mosenifar, M (ed.). "Hypoventilation Syndromes" . ... PMID 20304901 . ^ a b Trang H, Dehan M, Beaufils F, Zaccaria I, Amiel J, Gaultier C (2005). ... S.; Strandjord, T. P.; Badura, R. J.; Milstein, J. M. (1987). "Undine curse and neurocristopathy".
A number sign (#) is used with this entry because congenital central hypoventilation syndrome (CCHS) is most commonly caused by heterozygous mutation in the PHOX2B gene (603851) on chromosome 4p13. The disorder is also rarely caused by mutation in several other genes, including RET (164761), GDNF (600837), EDN3 (131242), and ASCL1 (100790). Evidence suggests that PHOX2B is the major disease-causing gene in isolated and syndromic CCHS (Amiel et al., 2003; Weese-Mayer et al., 2003). Haddad syndrome, in which CCHS is associated with Hirschsprung disease, is caused by heterozygous mutation in the ASCL1 gene (100790). Description Idiopathic congenital central hypoventilation syndrome, also known as 'Ondine's curse' (Deonna et al., 1974), is a rare disorder characterized by abnormal control of respiration in the absence of neuromuscular, lung or cardiac disease, or an identifiable brainstem lesion.
Haddad syndrome is a rare congenital disorder in which congenital central hypoventilation syndrome (CCHS), or Ondine syndrome, occurs concurrently with Hirschsprung disease (see these terms). Epidemiology Birth incidence of Ondine syndrome is 1 in 200,000 live-births and Hirschsprung disease occurs concurrently in 16% of cases. Clinical description Intestinal aganglionosis is more extensive, and the gender ratio is 1:1, unlike in classical Hirschsprung disease. Etiology Mutations in the PHOX2B gene are found in a significant number of patients with Haddad syndrome.
Congenital central hypoventilation syndrome (CCHS) is a disorder that affects normal breathing. People with this disorder take shallow breaths (hypoventilate), especially during sleep, resulting in a shortage of oxygen and a buildup of carbon dioxide in the blood. Ordinarily, the part of the nervous system that controls involuntary body processes (autonomic nervous system) would react to such an imbalance by stimulating the individual to breathe more deeply or wake up. This nervous system reaction is impaired in people with CCHS. They must be supported with a machine to help them breathe (mechanical ventilation) or a device that stimulates a normal breathing pattern (diaphragm pacemaker). Some affected individuals need this support 24 hours a day, while others need it only at night.
Hormone replacement therapy can also reduce the likelihood of osteoporosis. [9] Epidemiology [ edit ] It has been estimated that the incidence of Swyer syndrome is approximately 1 in 100,000 people. [10] Fewer than 100 cases have been reported as of 2018. [10] There are extremely rare instances of familial Swyer syndrome. [10] History [ edit ] Swyer syndrome was first described by G. I. M. Swyer in 1955 in a report of two cases. [10] References [ edit ] ^ Reference, Genetics Home. ... PMC 5877806 . PMID 27798415 . ^ Bomalaski, M. David (February 2005). "A practical approach to intersex" . ... MedlinePlus Genetics . ^ a b c d Banoth M, Naru RR, Inamdar MB, Chowhan AK (May 2018).
The vessels are obstructed by a coagulating current through the use of unipolar diathermy unit or by thermal cautery. [7] Research [ edit ] Reduction of neovascularization has been achieved in rats by the topical instillation of commercially available triamcinolone and doxycycline . [8] Some evidence exists to suggest that the Angiotensin II receptor blocker drug telmisartan will prevent corneal neovascularization. [2] Recent treatment developments include topical application of bevacizumab , an anti-VEGF. [9] References [ edit ] ^ a b c d e f g Abdelfattah N. S., Amgad M., Zayed A. A., Salem H., Elkhanany A. ... Ophthalmic Pearls : 35–36. ^ Riazi-Esfahani, M; Peyman, GA; Aydin, E; Kazi, AA; Kivilcim, M; Sanders, DR (August 2006).
. ^ Bara-Jimenez, W; Catalan, MJ; Hallett, M; Gerloff, C (1998). "Abnormal somatosensory homunculus in dystonia of the hand". ... Choosing music over meds, one man's quest to retrain his brain to overcome dystonia. https://www.youtube.com/watch?v=IpcXkV_ex8Y ^ Farias J, Yoshie M. Treatment efficacy in an ecologically valid neuropsycological treatment program of 120 professional musicians with focal dystonia , Galene Editions. ... Sources [ edit ] Tubiana, Raoul, Amadio, Peter C.; Medical Problems of the Instrumentalist Musician ; UK; Martin Dunitz (2000); 295-397 Rich, Robert F.; Mackin, Evelyn; Callahan, Anne; A. Lee Osterman; Terri M. Skirven; Schneider, Lawrence J. (2002).
Situs inversus is a condition in which the arrangement of the internal organs is a mirror image of normal anatomy. It can occur alone (isolated, with no other abnormalities or conditions) or it can occur as part of a syndrome with various other defects. Congenital heart defects are present in about 5-10% of affected people. The underlying cause and genetics of situs inversus are complex. Familial cases have been reported.
A rare, genetic, developmental defect during embryogenesis characterized by total mirror-image transposition of both thoracic and abdominal viscera across the left-right axis of the body. Congenital abnormalities, such as primary ciliary dyskinesia, Kartagener type, polysplenia syndrome, biliary atresia, congenital heart disease, and midgut malrotation, as well as vascular anomalies (e.g. absence of retrohepatic inferior vena cava, preduodenal portal vein, aberrant hepatic arterial anatomy) and malignancy, are frequently associated.
. ^ a b Shah, Ma; Salo-Mullen, E; Stadler, Z; Ruggeri, Jm; Mirander, M; Pristyazhnyuk, Y; Zhang, L (September 2012). ... PMID 18672140 . ^ Kriege, Mieke; Brekelmans, Cecile T. M.; Boetes, Carla; Besnard, Peter E.; Zonderland, Harmine M.; Obdeijn, Inge Marie; Manoliu, Radu A.; Kok, Theo; Peterse, Hans (2004-07-29).
A number sign (#) is used with this entry because hereditary diffuse gastric cancer (HDGC) and lobular breast cancer (LBC) are caused by heterozygous germline mutation in the E-cadherin gene (CDH1; 192090) on chromosome 16q22. Somatic mutation in the CDH1 gene has also been found in patients with sporadic diffuse gastric cancer and lobular breast cancer. Description Hereditary diffuse gastric cancer is an autosomal dominant cancer predisposition syndrome. Heterozygous CDH1 mutation carriers have a 70 to 80% lifetime risk of developing diffuse gastric cancer. In addition to gastric cancer, up to 60% of female mutation carriers develop lobular carcinoma of the breast, and some carriers may develop colorectal cancer.
Hereditary diffuse gastric cancer (HDGC) leads to an increased risk (predisposition) of developing a specific form of stomach cancer called diffuse gastric cancer . Women with HDGC also have an increased risk for lobular breast cancer . Cancers associated with HDGC generally occur at earlier ages than those seen in people who do not have a hereditary predisposition to cancer. HDGC is caused by genetic variants in the CDH1 gene and the CTNNA1 gene. It is inherited in an autosomal dominant pattern. Diagnosis of HDGC is based on the symptoms, family history, and may be confirmed by the results of genetic testing.
Hereditary diffuse gastric cancer (HDGC) is an inherited disorder that greatly increases the chance of developing a form of stomach(gastric) cancer. In this form, known as diffuse gastric cancer, there is no solid tumor. Instead cancerous (malignant) cells multiply underneath the stomach lining , making the lining thick and rigid. The invasive nature of this type of cancer makes it highly likely that these cancer cells will spread (metastasize ) to other tissues, such as the liver or nearby bones. Symptoms of diffuse gastric cancer occur late in the disease and can include stomach pain, nausea, vomiting, difficulty swallowing (dysphagia), decreased appetite, and weight loss.
Hereditary diffuse gastric cancer is a rare epithelial tumor of the stomach, characterized by the development of diffuse (signet ring cell) gastric cancer at a young age, associated with germline heterozygous mutations of CDH1 , MAP3K6 and CTNNA1 genes. In early stages it presents with non-specific and vague symptoms, in advanced stages it may cause nausea and vomiting, dysphagia, loss of appetite, abdominal mass or weight loss. Women have an increased risk of lobular breast cancer as well.
It is possible that bone marrow transplant may be helpful in delaying or correcting the neurological deterioration that occurs with I-Cell disease. [9] The Yash Gandhi Foundation is a US non-profit organization which funds research for I-Cell disease [10] References [ edit ] ^ " mucolipidosis II " at Dorland's Medical Dictionary ^ Plante M, Claveau S, Lepage P, et al. (March 2008). ... External links [ edit ] Classification D ICD - 10 : E77.0 ICD - 9-CM : 272.7 OMIM : 252500 MeSH : D009081 DiseasesDB : 29175 External resources eMedicine : ped/1150 GeneReviews : Mucolipidosis II mucolipidoses at NINDS — article derived from detail sheet available here I cell disease at NIH 's Office of Rare Diseases GeneReview/NIH/UW entry on Mucolipidosis II v t e Lysosomal storage diseases : Inborn errors of carbohydrate metabolism ( Glycoproteinoses ) Anabolism Dolichol kinase deficiency Congenital disorder of glycosylation Post-translational modification of lysosomal enzymes Mucolipidosis : I-cell disease (ML II) Pseudo-Hurler polydystrophy (ML III) Catabolism Aspartylglucosaminuria Fucosidosis mannosidosis Alpha-mannosidosis Beta-mannosidosis Sialidosis Schindler disease Other solute carrier family ( Salla disease ) Galactosialidosis v t e Disorders of translation and posttranslational modification Translation Ribosome : Diamond–Blackfan anemia FMR1 Fragile X syndrome Fragile X-associated tremor/ataxia syndrome Premature ovarian failure 1 Initiation factor : Leukoencephalopathy with vanishing white matter snRNP : Retinitis pigmentosa 33 Posttranslational modification Protein folding Alzheimer's disease Huntington's disease Creutzfeldt–Jakob disease chaperonins: 3-Methylglutaconic aciduria 5 Protein targeting I-cell disease Ubiquitin E1 : X-linked spinal muscular atrophy 2 E3 : Johanson–Blizzard syndrome Von Hippel–Lindau disease 3-M syndrome Angelman syndrome Deubiquitinating enzyme : Machado–Joseph disease Aneurysmal bone cyst Multiple familial trichoepithelioma 1 SUMO OFC10 Other Multiple sulfatase deficiency Hyperproinsulinemia Ehlers–Danlos syndrome 6
Recent investigational treatment therapies include high doses of Rituxan (Rituximab) and eculizumab, which are both humanized monoclonal antibodies that target B cell malignancies and prevent C5 complement cleavage, respectively. [12] References [ edit ] ^ a b c Nayer, Ali; Ortega, Luis M. (2014). "Catastrophic antiphospholipid syndrome: a clinical review" . ... PMID 20696282 . ^ MD Consult Retrieved on 2009-06-02 ^ Kazzaz, Nayef M.; McCune, W. Joseph; Knight, Jason S.
Catastrophic antiphospholipid syndrome (CAPS) is a rare form of antiphospholipid syndrome (APS). In CAPS multiple blood clots form throughout the body over a short period of time (usually within a week). CAPS is a medical emergency, as clots can cause life-threatening multi-organ failure. The cause of CAPS is unknown. A widely accepted explanation is that it is caused by a combination of gene mutations (making one more susceptible to CAPS) and an environmental trigger, such as an infection, trauma, or surgery.
A rare systemic autoimmune disease characterized by acute onset of life-threatening thromboses in three or more organs either simultaneously or within less than a week, in the presence of serum antiphospholipid antibodies (such as lupus anticoagulant, anticardiolipin antibodies, and anti-beta2-glycoprotein 1 antibodies), and with histopathological confirmation of small-vessel occlusion in at least one affected organ. The condition occurs in a small subset of patients with antiphospholipid syndrome, often precipitated by infection, trauma, or surgery.
The mechanism of this action was shown to be inhibition of DNA synthesis [10] Cannabinoids increase the life span of mice carrying Lewis lung tumors and decrease primary tumor size. [11] There are multiple modes of action. [12] References [ edit ] ^ Rashidi B, Yang M, Jiang P, Baranov E, An Z, Wang X, Moossa AR, Hoffman RM (2000-01-01). ... PMID 16250836 . ^ Portella G, Laezza C, Laccetti P, De Petrocellis L, Di Marzo V, Bifulco M (September 2003). "Inhibitory effects of cannabinoid CB1 receptor stimulation on tumor growth and metastatic spreading: actions on signals involved in angiogenesis and metastasis".