-
Ependymoma
Wikipedia
.; Kumar, Vinay; Fausto, Nelson; Nelso Fausto; Robbins, Stanley L.; Abbas, Abul K. (2005). "Ch. 28 The central nervous system".RELA, C11orf95, EPHB2, GDNF, RTBDN, BCL7C, RAB3A, ZNF668, TP53, NF2, IDH1, APC, TSC2, ERBB2, MSH2, MSH6, IFNG, MLH1, SETBP1, TSC1, PMS2, CDKN2A, YAP1, EGFR, MME, VEGFA, OLIG2, MKI67, MGMT, MDM2, MIB1, TP73, TTR, PTEN, MYCN, TNC, MAMLD1, L1CAM, EPB41L3, MEN1, FGFR1, NOTCH1, H3P10, EZHIP, NCL, ANXA1, CD274, ATRX, SST, CCND1, DAPK1, RASSF1, MIR17, NES, CDKN2B, MUC1, CD44, TNFRSF10C, PROM1, ABCB1, PDGFRA, SLC9A3R1, IL6, KIT, CASP8, FGFR3, DCX, HIC1, SOX10, SMARCB1, ARHGAP24, ETFA, GFAP, TNFSF10, SYP, GATD3A, LINC00899, MIR146B, IL18R1, ABCC8, ZNF148, KRT75, MIR449A, STC1, PSCA, CLDN5, TRERNA1, TERT, LOC110806263, TPR, TIMP3, LINC02210-CRHR1, THBS1, ABCG2, SIK1B, TSPAN4, TWIST1, UBE2N, GATD3B, TDG, VEGFB, ZEB1, SYT1, AZIN2, MVP, MIR10B, DNAJC15, ZMYND10, SHC3, TET2, FBXW7, BEX1, MIR10A, MIR100, LINC01194, HES4, GADL1, ZFHX4, SIK1, ARMC9, ESX1, C1orf194, TP53INP1, GLS2, CBY1, MIR330, KIF4A, EBI3, MIR135B, AKR1A1, MIR34C, TUBB3, MIR29A, LRIG3, HOXB13, MIR24-1, IPO7, MIR221, MIR19A, MXD4, MIR181C, MIR15A, DICER1, HEY2, STAT3, JAG1, SPINK1, EPO, MECOM, EZH2, FCGR1A, FHIT, FOXJ1, FLT1, GH1, GNAO1, HOXA9, HES1, HTC2, HTR1B, IDH2, IGF2, IGFBP2, IGFBP3, IGFBP5, IL1R1, CXCL8, JAG2, JAK2, ERCC1, EPB41, SOX11, S1PR3, AQP1, ASAH1, BDNF, BRAF, CALCA, CALCR, CALR, CAPS, CAV1, RUNX1, CD34, CD38, CD151, CDK6, CHI3L1, CCR7, CRHR1, CTNNB1, DAXX, DRD1, DRD2, KDR, LAMA2, RPSA, STMN1, PTPRS, RAC2, RAD51, ALK, RBL2, RFX3, RRM1, S100A1, S100A2, S100A4, S100A6, S100A12, S100B, SHH, SLC6A8, SMN1, SMN2, SNCA, SNCG, SOX4, SOX9, PTPRA, PTGS2, PLG, NFIC, LRP1, MAP2, MDM4, MET, CD99, ABCC1, MSH3, NELL2, NF1, NFKB1, PIK3CG, NGF, OTX2, PAX5, PAX6, PCNA, PDGFRB, PIK3CA, PIK3CB, PIK3CD, RARB

-
Caudal Regression Syndrome
Wikipedia
In 2012, Spencer West, a man with sacral agenesis and both legs amputated, climbed Mount Kilimanjaro using only his hands. [6] Society [ edit ] Notable cases [ edit ] Johnny Eck , American freak show performer Kenny Easterday , played a fictionalized version of himself in the film Kenny Kurt Fearnley , Australian wheelchair racer Bobby Martin , American footballer Jessica Rogers , American wheelchair racer and swimmer, founder and President of iSACRA (International Caudal Regression Syndrome Association) Rose Siggins , actress ( American Horror Story: Freak Show ) [7] Victoria Pendergast , first female Australian sit-skier at Winter Paralympics Rebecca Dubber , New Zealand para-swimmer and Rio 2016 Paralympic bronze medalist Kevin McKee , two-times olympic champion and two-times world champion in sledge hockey References [ edit ] ^ Sonek JD, Gabbe SG, Landon MB, Stempel LE, Foley MR, Shubert-Moell K (March 1990). "Antenatal diagnosis of sacral agenesis syndrome in a pregnancy complicated by diabetes mellitus".

-
Strain (Injury)
Wikipedia
PMID 26618057 . ^ a b Mulcahey, Mary K. (June 2020). "Sprains, Strains and Other Soft-Tissue Injuries" .
-
Exanthem
Wikipedia
PMID 11214144 . ^ Altman, Lawrence K (November 30, 1982). "THE DOCTOR'S WORLD" .HLA-B, MVK, C1QA, IL1RN, NOD2, GJA1, NLRP3, HLA-DRB1, CTLA4, PIK3R1, APOA1, SDHB, KRT5, TRPS1, KDSR, FGA, FBN1, NAXD, MEFV, EGFR, ERCC5, ERCC4, ERCC3, ERCC2, TNFRSF1B, TNFRSF1A, ENPP1, LBR, GJB3, GLUL, IL17RA, IGLL1, IGHM, PTPN22, IL6, RFXANK, CIITA, IKBKG, BLNK, SPINK5, POMP, ZAP70, HLCS, HLA-DPB1, HLA-DPA1, DNASE1L3, PDGFRA, LMBRD1, SMARCAD1, LRRC8A, MIF, CD28, RFX5, GJB4, BTK, BTD, RFXAP, KIT, LACC1, B2M, TEK, TCF3, ABCC6, SLC6A19, STAT3, SDHA, AK2, SDHC, RAG2, IL2RG, IL17F, LYZ, NLRC4, PRTN3, DCLRE1C, CLEC7A, PSMB4, PSMB8, PSMB9, COX4I2, IL17RC, TRAF3IP2, JAK3, CD79B, CD79A, NLRP12, RAG1, TP53, GPT, HLA-A, PTH1R, RUNX2, EGF, IFNG, NAT2, PIK3CA, CCR4, PTHLH, BRAF, TNFRSF11A, TNFSF11, PTH, PAEP, CD274, KRAS, IL5, CARD14, FAM20A, CSF3, GH1, HPGDS, TNFRSF8, AREG, ADNP, SYP, TSLP, TRIM21, SST, STAT1, SPRTN, TNF, CDAGS, HAVCR2, TGFB1, IL33, MIR126, MUC16, MIR31, MIR146A, TGFA, IL21, TIRAP, RBM45, UNC13D, IFIH1, ASPN, ENOSF1, ODAM, PGRMC1, GNLY, MRPL28, SUB1, SAE1, ADAMTS1, SDS, SATB2, RECQL4, DIRAS3, NAE1, IL1RL2, SMUG1, ABCB11, NXT1, SMN2, IL22, SIRT6, ARID4B, XRCC1, UBASH3A, TPI1, CCL27, SOD1, A1BG, SMN1, SMARCB1, CYBB, CYP2C9, CYP2E1, CYP27B1, NQO1, DLX4, DMP1, DSG1, EEC1, EIF4EBP1, ENO1, EREG, ESAT, GATA1, GNRH1, HMGB1, HSPA8, IAPP, IDH1, CTSK, CSTA, MAPK14, SERPING1, ACTB, AKT1, ALPP, AMBN, AR, RHOH, BCL6, BST2, CASP3, CRYZ, CD1A, CD34, CD44, CD68, CDKN2B, CFTR, CKB, CPA1, IDH2, IFNA1, IFNA13, PTEN, TNFRSF11B, PDCD1, CFP, ABCB1, PHB, PIK3CB, PIK3CD, PIK3CG, PTGS2, NPHS1, PLAAT4, SERPINB4, CCL11, CCL17, CCL22, SDHD, SLC1A3, SLC22A1, OPA1, NM, CCN1, LDHB, IL1B, IL2, IL4, CXCL8, IL9, IL10, IL17A, CXCL10, LGALS3, MTRR, LGALS9, LHCGR, ADAM11, MECP2, KMT2A, MMP2, MMP9, MPO, H3P9

-
Thunderstorm Asthma
Wikipedia
PMID 18587040 ^ Forouzan A, Masoumi K, Haddadzadeh Shoushtari M, Idani E, Tirandaz F, Feli M, Assarehzadegan MA, Asgari Darian A.

-
Flock Worker's Lung
Wikipedia
. ^ a b c d Eschenbacher, W. L.; Kreiss, K.; Lougheed, M. D.; Pransky, G. S.; Day, B.; Castellan, R.

-
Paget–schroetter Disease
Wikipedia
.; Wells, Philip S.; Gould, Michael K. (2012-02-01). "Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines" .

-
Hypertensive Retinopathy
Wikipedia
See also [ edit ] Hypertensive crisis List of systemic diseases with ocular manifestations Ophthalmology Optometry References [ edit ] ^ a b Bhargava, M; Ikram, M K; Wong, T Y (2011). "How does hypertension affect your eyes?"TMEM127, MDH2, SDHB, SDHA, RET, VHL, SLC25A11, KIF1B, MAX, SDHD, SDHAF2, HSD11B2, GDNF, FH, DLST, SDHC, ANO1, CTRL, DLD, PSMD9, PSMB10, IKBKB, EDN1, RAC1

-
Hidradenitis
Wikipedia
McGraw-Hill. ISBN 0-07-138076-0 . ^ Sellheyer K, Krahl D (July 2005). " " Hidradenitis suppurativa" is acne inversa!
-
Ulnar Collateral Ligament Injury Of The Thumb
Wikipedia
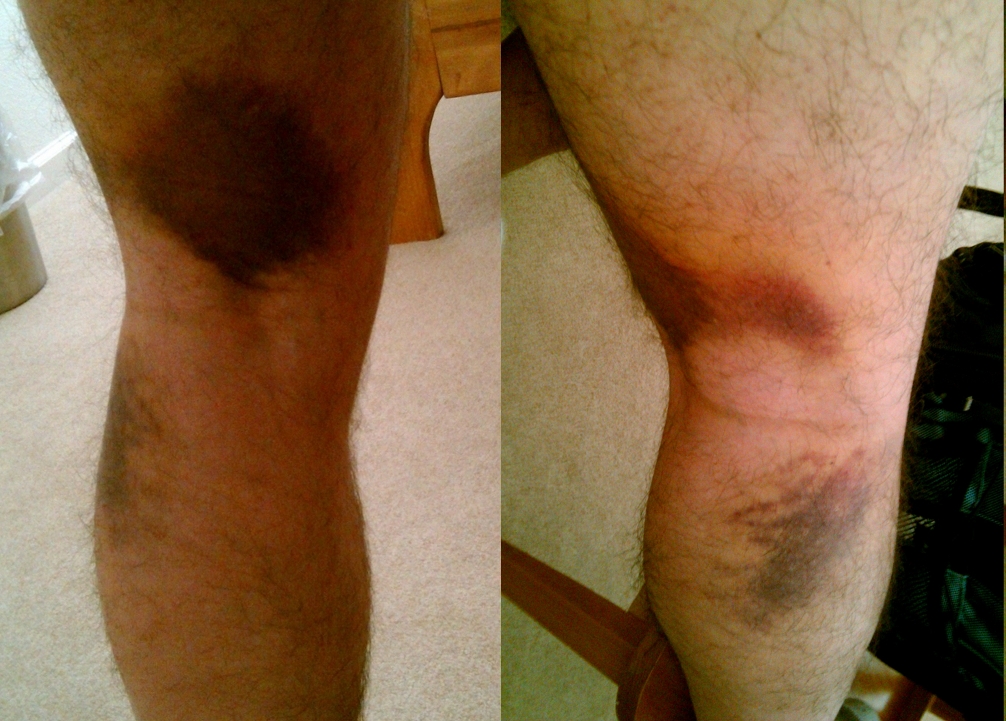
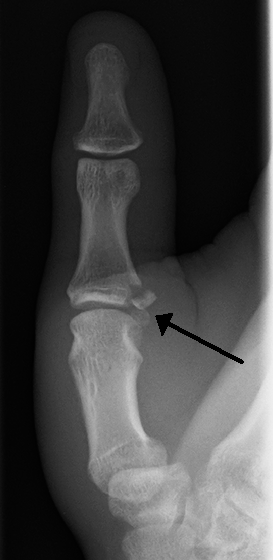
Sports injuries accounted for most of the remaining injuries, with only 2.4% acquired as a result of skiing injuries. [6] Treatment [ edit ] A post-operative photo of repair of a complete rupture of the ulnar collateral ligament. Note the K-wire to brace the joint. The ulnar collateral ligament is an important stabilizer of the thumb.
-
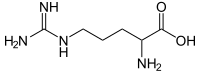
Argininemia
Wikipedia
External links [ edit ] Classification D ICD - 10 : E72.2 ICD - 10-CM : E72.21 OMIM : 207800 MeSH : D020162 DiseasesDB : 29677 External resources eMedicine : ped/132 GeneReviews : Arginase Deficiency Orphanet : 90 Scholia has a topic profile for Argininemia . v t e Inborn error of amino acid metabolism K → acetyl-CoA Lysine /straight chain Glutaric acidemia type 1 type 2 Hyperlysinemia Pipecolic acidemia Saccharopinuria Leucine 3-hydroxy-3-methylglutaryl-CoA lyase deficiency 3-Methylcrotonyl-CoA carboxylase deficiency 3-Methylglutaconic aciduria 1 Isovaleric acidemia Maple syrup urine disease Tryptophan Hypertryptophanemia G G→ pyruvate → citrate Glycine D-Glyceric acidemia Glutathione synthetase deficiency Sarcosinemia Glycine → Creatine : GAMT deficiency Glycine encephalopathy G→ glutamate → α-ketoglutarate Histidine Carnosinemia Histidinemia Urocanic aciduria Proline Hyperprolinemia Prolidase deficiency Glutamate / glutamine SSADHD G→ propionyl-CoA → succinyl-CoA Valine Hypervalinemia Isobutyryl-CoA dehydrogenase deficiency Maple syrup urine disease Isoleucine 2-Methylbutyryl-CoA dehydrogenase deficiency Beta-ketothiolase deficiency Maple syrup urine disease Methionine Cystathioninuria Homocystinuria Hypermethioninemia General BC / OA Methylmalonic acidemia Methylmalonyl-CoA mutase deficiency Propionic acidemia G→ fumarate Phenylalanine / tyrosine Phenylketonuria 6-Pyruvoyltetrahydropterin synthase deficiency Tetrahydrobiopterin deficiency Tyrosinemia Alkaptonuria / Ochronosis Tyrosinemia type I Tyrosinemia type II Tyrosinemia type III / Hawkinsinuria Tyrosine → Melanin Albinism : Ocular albinism ( 1 ) Oculocutaneous albinism ( Hermansky–Pudlak syndrome ) Waardenburg syndrome Tyrosine → Norepinephrine Dopamine beta hydroxylase deficiency reverse: Brunner syndrome G→ oxaloacetate Urea cycle / Hyperammonemia ( arginine aspartate ) Argininemia Argininosuccinic aciduria Carbamoyl phosphate synthetase I deficiency Citrullinemia N-Acetylglutamate synthase deficiency Ornithine transcarbamylase deficiency / translocase deficiency Transport / IE of RTT Solute carrier family : Cystinuria Hartnup disease Iminoglycinuria Lysinuric protein intolerance Fanconi syndrome : Oculocerebrorenal syndrome Cystinosis Other 2-Hydroxyglutaric aciduria Aminoacylase 1 deficiency Ethylmalonic encephalopathy Fumarase deficiency Trimethylaminuria v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
-
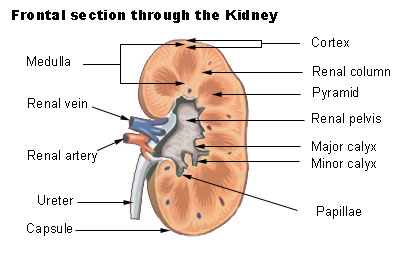
Renal Papillary Necrosis
Wikipedia
. ^ Greenberg, Arthur; Cheung, Alfred K. (2005-01-01). Primer on Kidney Diseases .
-
Rotor Syndrome
Wikipedia
Genes, SLCO1B1 and SLCO1B3 that result in complete functional deficiencies of both protein products (OATP1B1 and OATP1B3, respectively), are also present. [ citation needed ] Rotor syndrome is largely a diagnosis of exclusion. [2] Serological abnormalities in Rotor syndrome only include elevated total serum bilirubin (typically elevated between 2 to 5 mg/dL but may be as high as 20 mg/dL). [2] Most of the time, alanine aminotransferase , aspartate aminotransferase , gamma-glutamyl transferase , and alkaline phosphatase levels are normal, but mild elevations can be seen. [2] If any of these lab values are markedly elevated, investigation for other, more serious conditions is warranted. [2] Imaging studies cannot diagnose Rotor syndrome but can help rule out other diseases that cause hyperbilirubinemia . [2] For example, ultrasound of the liver and the biliary tree can help investigate the causes of extra-hepatic biliary obstruction . [2] The gallbladder is visualized on oral cholecystography in Rotor syndrome while it is not visualized in Dubin Johnson syndrome. [2] Ultimately, the best method of diagnosing the disease is the analysis of urine coproporphyrin excretion. [2] The total urine coproporphyrin excretion in Rotor syndrome has a 2- to 5-fold elevation, with 65% constituting coproporphyrin I. [2] Treatment [ edit ] To treat the jaundice, phenobarbital is normally used. [ citation needed ] Rotor syndrome is a benign disease requiring no treatment. [2] Jaundice is a lifelong finding, but the disease is not associated with morbidity or mortality, and life expectancy is not affected. [2] Most individuals with Rotor syndrome are born to consanguineous couples and its diagnosis may coincidently identify consanguinity. [2] Distinguishing Rotor syndrome from other more serious disorders is important to avoid unnecessary workup and interventions. [2] It is also critical to reassure and calm patients or family members of patients with Rotors syndrome that the condition is benign. [2] Eponym [ edit ] Rotor syndrome is named after the Filipino internist Arturo Belleza Rotor (1907–1988). [8] See also [ edit ] Jaundice Bilirubin metabolism Gilbert's syndrome Crigler–Najjar syndrome References [ edit ] ^ a b Online Mendelian Inheritance in Man (OMIM): 237450 ^ a b c d e f g h i j k l m n o p q r s t Kumar, A; Mehta, D (2020), "article-36575", Rotor Syndrome , This book is distributed under the terms of the Creative Commons Attribution 4.0 International License ( http://creativecommons.org/licenses/by/4.0/ ), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, a link is provided to the Creative Commons license, and any changes made are indicated., Treasure Island (FL): StatPearls Publishing, PMID 30335339 , retrieved 2020-07-17 ^ a b Wolkoff AW, Wolpert E, Pascasio FN, Arias IM (February 1976).

-
Aneurysm Of Sinus Of Valsalva
Wikipedia
. ^ Vural KM, Sener E, Taşdemir O, Bayazit K (July 2001). "Approach to sinus of Valsalva aneurysms: a review of 53 cases" .
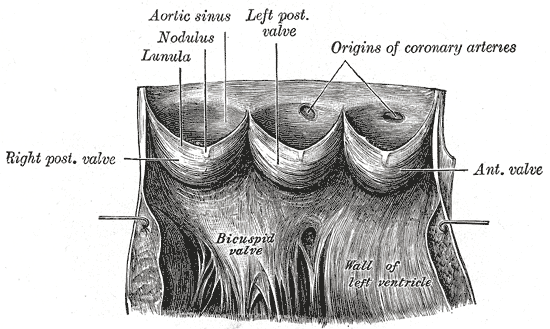
-
Transient Hyperammonemia Of The Newborn
Wikipedia
Retrieved November 12, 2012 . ^ a b c d e f g h i j k l m n o p Krishnan, L.; Diwakar, K.K.; Patil, P.; Bhaskaranand, N. (1996).
-
Phosphorus Deficiency
Wikipedia
., Bekjarov, G., Dospatliev, L., Ivanov, & K., Zaprjanova, P. (2010). ICP determination of phosphorus in soils and plants.

-
Stasis Papillomatosis
Wikipedia
Obtenido de Clinical Key . [ full citation needed ] ^ a b c d e Sato T, Katagiri K, Itami S, Takayasu S (May 1996). "A case of stasis papillomatosis associated with psoriasis vulgaris".
-
Epiploic Appendagitis
Wikipedia
BMC Surgery 7:11. ^ a b c d e f g h i Singh, Ajay K.; Gervais, Debra A.; Hahn, Peter F.; Sagar, Pallavi; Mueller, Peter R.; Novelline, Robert A. (2005-11-01).

-
Comedocarcinoma
Wikipedia
. ^ a b "Understanding Your Pathology Report: Ductal Carcinoma In Situ (DCIS)" . www.cancer.org . Retrieved 2020-04-23 . ^ Pai, K., Baliga, P., & Shrestha, B. L. (2013).
-
Phantom Vibration Syndrome
Wikipedia
Retrieved 2019-02-11 . ^ a b c d e f g h i j k Deb A (2014). "Phantom vibration and phantom ringing among mobile phone users: A systematic review of literature".














