-
Hidradenitis Suppurativa
Wikipedia
This theory includes most of the following potential indicators: [7] Post-pubescent individuals [8] Blocked hair follicles or blocked apocrine sweat glands Excessive sweating Androgen dysfunction Genetic disorders that alter cell structure Patients with more advanced cases may find exercise intolerably painful, which may increase the rate of obesity among sufferers. The historical understanding of the disease suggests dysfunctional apocrine glands [9] or dysfunctional hair follicles, [10] possibly triggered by a blocked gland, which creates inflammation , pain , and a swollen lesion .NCSTN, PSENEN, NLRP3, MEFV, NOD2, PSTPIP1, GJB2, PSEN1, IL17A, TNF, HYOU1, KDF1, IL1B, IL1A, IFNG, CRP, IL23A, IL10, IL22, CXCL8, PAPPA, IL6, IL20, GLI3, IL36RN, IL17B, DCD, IL37, IL13, AGO1, YME1L1, ADIPOQ, IL32, ACAD8, ADM, SND1, AGO2, C5AR2, SULT1B1, KRT20, IL26, RETN, ELOVL7, IL1F10, MTDH, RBM45, TET3, IL1RL2, SAA1, TLR4, IFNA1, BCL2, CAMP, MS4A1, CD27, CHI3L1, CTNNB1, CTNND1, EPHB2, ERBB4, HLA-A, HLA-DRB1, IDH1, IDH2, IDH3B, IFNA13, TIE1, IL12RB1, ITGAL, ITGB2, KLRB1, LCN2, CYP4F3, NHS, P2RX7, PDE4A, ANXA5, SAA2, SULT1E1, TARBP2, TGFB1, MIR21

-
Mood Disorder
Wikipedia
These characteristics would be difficult to understand if depression were a dysfunction. [70] A depressed mood is a predictable response to certain types of life occurrences, such as loss of status, divorce, or death of a child or spouse. ... CS1 maint: archived copy as title ( link ) ^ a b Pieczenik, Steve R, Neustadt, John (2007). "Mitochondrial dysfunction and molecular pathways of disease". ... PMID 17239370 . ^ "Mitochondrial dysfunction and bipolar disorder" . 29 September 2017. ... Retrieved 1 October 2017 . Mitochondrial dysfunction and bipolar disorder ^ Scainiab, Giselli, Rezinc, Gislaine, Carvalhod, Andre, Streckb, Emilio L, Berkef, Michael, Quevedo, João (2016). "Mitochondrial dysfunction in bipolar disorder: Evidence, pathophysiology and translational implications".SLC6A4, NR3C1, POLG, ADCY8, DRD2, NCAM1, CREB1, COMT, BDNF, HTR2A, CRH, CLOCK, DISC1, MAOA, TH, HTR1A, FKBP5, P2RX7, DRD4, TPH1, CACNA1C, TPH2, WFS1, NPY, GSK3B, TNF, TAC1, CRHR1, HTR2C, NTRK2, GPR50, ESR1, OPRK1, PRL, DBH, ABO, HP, GABRA1, SLC6A3, SIRT2, MTHFR, DLG4, GRIN2A, DTNBP1, PDYN, AKT1, DRD3, PROKR2, MCHR1, HTR5A, AVPR1B, GNB3, ZNF804A, FGF2, DAOA, S100B, RELN, MAOB, FMR1, GAL, HCRTR1, NTF3, DLG3, IMPA2, GRM3, GRM2, GAD1, GRIK2, MAP2, GAD2, PROK2, HTR3A, FAAH, IL1RN, DISC2, HTR6, CYP2D6, TACR1, SLC6A2, ARNTL, SMS, CRHBP, PLA2G1B, TIMELESS, SERPINA1, ATP2A2, CRHR2, AR, GABRB2, GABRA6, GABRA5, CRTC1, SLC12A5, MAGEL2, SIGMAR1, RARA, VPS13A, ADRA2A, VIP, GABRD, SIRT1, VGF, SAT1, FEV, ANK3, HTR4, SLC1A2, TSNAX, VEGFA, TSNAX-DISC1, SLC5A7, HES6, GPRC5D, HPGDS, ADRB1, GCH1, ARRB2, TSPAN8, IL9R, SPR, FZD3, HCN4, CRYZ, TBX1, NTSR1, DAO, HIF1A, TGFB1, NTRK3, TGOLN2, NQO1, PAWR, NPY2R, HSPA4, DNMT1, DNMT3B, EDN1, PNOC, HTR1B, ADORA2A, ADCY7, MYO5B, PFKL, SST, RNPS1, CHRM2, CHRM3, ABCG1, FTO, SSTR3, SERTAD1, HSP90AA1, OXT, HTR7, NFKB1, TBC1D25, NTS, HDAC4, RGS6, TNFSF13B, PFKFB3, ERDA1, BSCL2, KAT2B, PMCH, PPARG, PRNP, LDHA, S100A10, KCNK9, PTGS2, HDAC2, SMOX, GAP43, FOSB, DUSP6, GLUL, DCTN1, CACNA1D, AMBP, HDAC1, ADRA2C, ADRA1B, ADCY3, HTT, KCTD12, HCN1, LINC02210-CRHR1, ERBB4, PDE4B, NEGR1, DCLK2, LINC01618, CDH8, MAD1L1, DPY19L3, PHF2, AREL1, EGFLAM, KLHL29, PAFAH1B1, CSE1L, RABGAP1L, SPSB4, NR1H3, CSTF3, NAV3, DAG1, BAIAP2, DGKG, KLHDC8B, WNT3, SSPN, SLC44A5, TCF4, LINC02040, GNG12-AS1, NCAM1-AS1, C6orf99, APBB2, UBA7, MAPT-AS1, RERE, LINC00461, SMIM4, RAPSN, RAB27B, C5orf17, PTPRD, PTH2R, VWC2L, VRK2, LINC01122, FAM228B, EYS, KANSL1, MYO1H, DCC, CREB3L1, ATAD2B, MAPT, EXD2, C11orf49, TNC, SOX6, PPP4R3A, NCOA5, ARHGAP15, YLPM1, CELF4, SORCS3, ZHX3, LMOD1, FSTL4, PLCL2, DPP10, KAZN, BBX, CAMTA1, NUP160, GPM6A, PBRM1, FYB1, TTC12, DDB2, LINGO1, ATP5MD, DELEC1, ARPP21, ARFGAP2, BICRA, KIAA1109, PRR16, NLK, TFAP2D, EMCN, CSNK1G1, IGLV10-54, BHLHE41, TMEM106B, ADAMTS6, BAZ1A, RBFOX1, GABBR1, PON1, ACE, IL6, PER3, CRP, IL1B, IGFBP2, LOC110806262, STIN2-VNTR, EPO, IL10, REM1, NR3C2, APOE, C1QL1, KL, TMED2, TPO, TPPP, P2RX5-TAX1BP3, TEMPS, BICC1, PPP1R9B, OPN4, CHP1, NLRP3, MAFD6, YWHAZ, NRSN1, HOMER1, MAFD4, PLB1, P2RX6, DGKH, P2RX2, FXR1, SMC2, H3P19, P2RX3, DST, PIK3CD, PIK3CB, PIK3CA, CD9, CNR1, P2RY2, P2RY1, P2RX5, P2RX4, CRY2, P2RX1, CTNNB1, DNASE1L3, DRD1, NOS1, MAFD2, MAFD1, FKBP4, GLP1R, GPER1, GRIK4, GRIN2B, HCRT, PIK3CG, GRM7, AVP, ADARB1, PVALB, MIR18A, EIF4EBP1, GABPA, FTL, FOS, FOXO3, ADCY9, PINK1, UBE2Z, FGF9, DMTN, MAPKAP1, LIN28A, SLC17A6, ADRA1A, ADRA2B, AGMAT, CAMKMT, RABEP2, EGF, PPP1R2C, PNPLA3, ZNF34, ADRB2, EDNRA, NDRG2, GABRA3, ACKR3, GLI2, LINC02605, GRM5, ACTB, GRIA3, GRIA1, PPP1R12C, GPR42, RN7SL263P, ADCY1, GLO1, GHSR, AP2B1, PLXNA3, GFAP, ATF7IP, CNDP2, GDNF, USE1, LMO3, GPRC5C, GCHFR, GCG, TMPRSS13, DPYD, PPP1R1B, CECR, CD44, KRIT1, CCKAR, CCK, CRTC2, CAT, OPN1SW, MDD1, MIR17HG, C9orf72, CALR, CALM3, IS1, CALM2, DAOA-AS1, CALM1, CALCA, TAAR6, NANOS3, MCIDAS, TSPO, BRS3, BRCA1, CDSN, OSR1, BPI, CHGB, APLNR, DMD, DLX4, DDC, SHANK3, RGS8, LMLN, CYP19A1, WASHC1, CSNK1E, ANGPT1, CSF2, PDIA3, CAMK2N2, SLC25A4, APOH, STS, CRY1, WASH6P, CREBBP, CP, ATP5F1A, BDNF-AS, SYN2, VSX1, GSTM1, ORM1, PLCB4, PLA2G4A, ST8SIA2, PITX2, ELP1, CCN6, PIK3R2, APLN, PIK3R1, ARHGEF7, CBFA2T2, LPAR2, HTR3B, ABCB1, ADIPOQ, ABCG2, EIF2AK3, PGM1, GDF15, NR1D1, PGD, PAX6, SEC24C, HDAC9, KEAP1, CCS, HDAC6, FOSL1, RASSF7, POU3F1, SLC18A2, SULT2A1, STAR, SSTR5, TDGF1P3, TFRC, SSTR4, SSB, THBS3, SOD2, TM7SF2, SOD1, SOAT1, TRPM2, PTPA, SEMG1, TTN, TYR, CCL11, CCL2, S100A12, VIPR2, PTGS1, VTN, PSMB6, PRKCZ, YY1, TOM1, TUBA1B, GSTT1, RAPGEF3, INSRR, PPP1R13B, IL7, SIRT3, IL4, IL2, IGHG1, ATP2C1, AGO2, IFNG, IFNA13, PCDH17, IFNA1, PCLO, DLL1, HTR1F, IGLV3-25, HSD11B2, SETD2, HRAS, HPX, SLC40A1, VAMP7, HPD, HLA-C, CRYL1, HINT1, ITGA2B, ITIH1, PLCB1, NGF, NUP98, DEAF1, NTRK1, PDLIM5, NRAS, CXCR6, NPAS2, NRG3, NOTCH4, NOS3, PPARGC1A, UTS2, METAP2, ITIH3, RAPGEF4, NFE2L2, MST1, MSMB, MAS1, LTBP3, IGSF9B, LEP, LBP, TBC1D9, STAB1, LAMC2, ABAT

-
Spinocerebellar Ataxia Type 36
GeneReviews
The diagnosis is suspected based on clinical findings in the absence of primary causes of cerebellar dysfunction. It is supported by a family history consistent with autosomal dominant inheritance, which can include simplex cases (i.e., a single occurrence in a family). ... However, in advanced disease the voice acquires a mixed quality with associated bulbar and/or pseudobulbar dysfunction. Appendicular cerebellar signs are also present in virtually all patients, manifesting as dysmetria and dysdiadochokinesis. ... Examination by an otolaryngologist and audiologist, with emphasis in a comprehensive characterization of degree and anatomic level of hearing dysfunction. Clinical genetics consultation and genetic counseling Additional brain MRI is not necessary following the diagnosis of SCA36; however, it can be used for complementary follow-up evaluation.
-
Hypoalphalipoproteinemia, Primary, 2
OMIM
Description Primary hypoalphalipoproteinemia-2 characterized by dysfunctional apoA-I production, resulting in undetectable levels of apoA-I in serum and in markedly low levels of serum high density lipoprotein cholesterol (HDL-C), is generally an autosomal recessive disorder associated with extensive atherosclerosis, xanthomas, and corneal opacities (summary by Tanaka et al., 2018).
-
Myopathy Due To Myoadenylate Deaminase Deficiency
OMIM
The findings suggested that disruption of the purine nucleotide cycle due to myoadenylate deaminase deficiency can result in marked alterations in ATP content of muscle, and that the changes could account for muscle dysfunction. Shumate et al. (1980) reported an 18-month-old girl referred for delayed motor and speech development.
- Cranioectodermal Dysplasia 2 OMIM
- Congenital Disorder Of Glycosylation, Type Ie OMIM
-
Orotic Aciduria
OMIM
INHERITANCE - Autosomal recessive GROWTH Other - Failure to thrive (in some patients) CARDIOVASCULAR Heart - Atrial septal defect (in 1 patient) - Ventricular septal defect (in 1 patient) GENITOURINARY - Orotic acid urinary obstruction NEUROLOGIC Central Nervous System - Developmental delay (in some patients) METABOLIC FEATURES - Orotic aciduria HEMATOLOGY - Megaloblastic anemia - Low to normal reticulocyte count - Anisocytosis - Poikilocytosis - Hypochromia - Platelet count normal IMMUNOLOGY - T-cell dysfunction, variable (in some patients) LABORATORY ABNORMALITIES - Orotic aciduria - Orotic acid crystalluria - Hematuria MOLECULAR BASIS - Caused by mutation in the uridine monophosphate synthetase gene (UMPS, 613891.0001 ) ▲ Close

- Cone-Rod Dystrophy 6 OMIM
-
Cleft Palate With Or Without Ankyloglossia, X-Linked
OMIM
The proband had a submucous cleft palate, ankyloglossia, speech and language delay, and left-sided eustachian tube dysfunction. His carrier mother had ankyloglossia, which was widely present in the extended family; affected males in the family also had submucous or soft palate cleft.
-
Papillon-Lefevre Syndrome
OMIM
They found several other reports of this association and concluded that liver abscess is an important complication of neutrophil dysfunction in PLS. Toomes et al. (1999) summarized the clinical features of Papillon-Lefevre syndrome.CTSC, ABO, IGFALS, SOD1, SPG7, TP53, FIG4, CCL3L3, C9orf72, ALS2, CASZ1, TBK1, ERLIN2, FASTK, SLC35A1, GAL3ST1, DPYS, ELANE, SPAST, CAMP, CCL3L1, CCL3, PLS1, CXCL10, IL2, DDIT3, HLA-DRB1, GZMB, CST12P

-
Thrombocythemia 1
OMIM
Platelets from 2 of these patients showed dysfunction, including failure to aggregate or release serotonin in response to concentrations of epinephrine that aggregated platelets of normal controls.JAK2, CALR, SH2B3, MPL, THPO, IFNA2, TET2, TP53, TGFB1, PDGFA, FGF2, PDGFB, MYB, BCR, ABL1, CD34, CD177, ASXL1, PRB1, SOAT1, EPO, IFNA13, IFNA1, STAT5A, IDH2, STAT5B, VEGFA, LINC01152, AR, SRSF2, GATA1, F5, F2, SELP, IDH1, STAT3, DERL1, NFE2, IL6, SOCS3, BCL2, F3, HPSE, STAT1, CXCL8, TFPI, ITGAM, ITGB3, SF3B1, KIT, PTX3, LCN2, MDM2, PGF, SELE, MMP9, TERT, MVD, MYC, NOS3, SOCS1, HMGA2, HBS1L, RN7SL263P, FLI1, CSF2, CBL, EPOR, CD63, CTNNB1, G6PD, EVPL, EZH2, BAX, CRP, CSF3R, LRPPRC, PIAS3, WDR4, HLA-B, BAK1, CHEK2, CCND1, DAAM1, MTUS2, ARSA, DKK1, HDAC9, HDAC6, NAAA, TLR4, BNIP3L, U2AF1, UCP2, BID, CXCR4, LAP, BDNF, SLC14A2, CNTNAP1, BCL2L2, DLK1, SOCS2, BCL2L1, USP14, MYOM2, GDF15, DKK3, RABGEF1, IL37, MIR146B, CYGB, ANXA5, MIR125A, MIR143, MIR203A, MIR221, MIR490, GGTLC5P, THBS1, GGTLC3, GGT2, GGTLC4P, BCL2L2-PABPN1, MIR4639, AK6, AKT1, NLRP3, MLIP, HAVCR2, PRAM1, SETD2, IL22, PTOV1, TERF2IP, ARG2, BCOR, FASLG, MTUS1, HAMP, ACE2, APOA1, HDAC11, FIP1L1, QTRT1, SESN2, CALB2, CD69, CASP9, RUNX1T1, ITGB2, FLT1, JAK1, FLII, FCGR2A, LDHA, LDHC, LGALS3, LIG3, SMAD4, MCAM, FANCB, MFGE8, ATXN3, MMP2, ITGA2B, IL10, CXCR2, IFNG, HIF1A, HFE, IFI27, SERPIND1, H2AX, CXCL1, IGF1, CXCR1, IGF2, IGH, IL3, NR3C1, IL6ST, GGT1, COX1, MTHFR, ERCC2, SELENOP, S100A9, S100A12, ACSM3, CXCL12, CHIT1, CHI3L1, AKR1C4, REN, SLC14A1, CDR1, SPP1, CDH13, HSPA4, CD14, S100A8, RARS1, EDN1, SERPINE1, NFATC2, ACE, ADM, NRAS, NTRK1, PAEP, PCNA, RAP1A, PDGFRA, CPB1, PROC, PROS1, PTGS1, CISH, NM

-
Schizophrenia 1
OMIM
Neither individual had neurologic dysfunction or mental retardation. The woman who was mother and sister of the affected men was phenotypically normal; her chromosomes showed a balanced translocation t(1;5)(q32.3;q13.3q11.2).
-
Löffler's Syndrome
Wikipedia
External links [ edit ] Classification D ICD - 10 : J82 ICD - 9-CM : 518.3 MeSH : D011657 DiseasesDB : 7580 External resources MedlinePlus : 000105 eMedicine : ped/1322 v t e Diseases of the respiratory system Upper RT (including URTIs , common cold ) Head sinuses Sinusitis nose Rhinitis Vasomotor rhinitis Atrophic rhinitis Hay fever Nasal polyp Rhinorrhea nasal septum Nasal septum deviation Nasal septum perforation Nasal septal hematoma tonsil Tonsillitis Adenoid hypertrophy Peritonsillar abscess Neck pharynx Pharyngitis Strep throat Laryngopharyngeal reflux (LPR) Retropharyngeal abscess larynx Croup Laryngomalacia Laryngeal cyst Laryngitis Laryngopharyngeal reflux (LPR) Laryngospasm vocal cords Laryngopharyngeal reflux (LPR) Vocal fold nodule Vocal fold paresis Vocal cord dysfunction epiglottis Epiglottitis trachea Tracheitis Laryngotracheal stenosis Lower RT / lung disease (including LRTIs ) Bronchial / obstructive acute Acute bronchitis chronic COPD Chronic bronchitis Acute exacerbation of COPD ) Asthma ( Status asthmaticus Aspirin-induced Exercise-induced Bronchiectasis Cystic fibrosis unspecified Bronchitis Bronchiolitis Bronchiolitis obliterans Diffuse panbronchiolitis Interstitial / restrictive ( fibrosis ) External agents/ occupational lung disease Pneumoconiosis Aluminosis Asbestosis Baritosis Bauxite fibrosis Berylliosis Caplan's syndrome Chalicosis Coalworker's pneumoconiosis Siderosis Silicosis Talcosis Byssinosis Hypersensitivity pneumonitis Bagassosis Bird fancier's lung Farmer's lung Lycoperdonosis Other ARDS Combined pulmonary fibrosis and emphysema Pulmonary edema Löffler's syndrome / Eosinophilic pneumonia Respiratory hypersensitivity Allergic bronchopulmonary aspergillosis Hamman-Rich syndrome Idiopathic pulmonary fibrosis Sarcoidosis Vaping-associated pulmonary injury Obstructive / Restrictive Pneumonia / pneumonitis By pathogen Viral Bacterial Pneumococcal Klebsiella Atypical bacterial Mycoplasma Legionnaires' disease Chlamydiae Fungal Pneumocystis Parasitic noninfectious Chemical / Mendelson's syndrome Aspiration / Lipid By vector/route Community-acquired Healthcare-associated Hospital-acquired By distribution Broncho- Lobar IIP UIP DIP BOOP-COP NSIP RB Other Atelectasis circulatory Pulmonary hypertension Pulmonary embolism Lung abscess Pleural cavity / mediastinum Pleural disease Pleuritis/pleurisy Pneumothorax / Hemopneumothorax Pleural effusion Hemothorax Hydrothorax Chylothorax Empyema/pyothorax Malignant Fibrothorax Mediastinal disease Mediastinitis Mediastinal emphysema Other/general Respiratory failure Influenza Common cold SARS Coronavirus disease 2019 Idiopathic pulmonary haemosiderosis Pulmonary alveolar proteinosis

-
Macular Degeneration, Age-Related, 13
OMIM
Seddon et al. (2013) found that 7.8% of ARMD cases compared to 2.3% of controls were carriers of rare missense CFI variants (odds ratio = 3.6; p = 2 x 10(-8)). There was a preponderance of dysfunctional variants in cases compared to controls.
-
Gastric Volvulus
Wikipedia
PMID 9074918 . v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum
-
Mental Retardation With Language Impairment And With Or Without Autistic Features
OMIM
In infancy, he had failure to thrive associated with oromotor dysfunction and excessive drooling. He showed delayed psychomotor development, with walking at age 25 months and a notable delay in speech and language acquisition with articulation difficulties.
-
Caplan's Syndrome
Wikipedia
External links [ edit ] Classification D ICD - 10 : J99.0 M05.1 ICD - 9-CM : 714.81 MeSH : D002205 DiseasesDB : 1961 SNOMED CT : 398640008 External resources MedlinePlus : 000137 Patient UK : Caplan's syndrome 00057 at CHORUS v t e Diseases of the respiratory system Upper RT (including URTIs , common cold ) Head sinuses Sinusitis nose Rhinitis Vasomotor rhinitis Atrophic rhinitis Hay fever Nasal polyp Rhinorrhea nasal septum Nasal septum deviation Nasal septum perforation Nasal septal hematoma tonsil Tonsillitis Adenoid hypertrophy Peritonsillar abscess Neck pharynx Pharyngitis Strep throat Laryngopharyngeal reflux (LPR) Retropharyngeal abscess larynx Croup Laryngomalacia Laryngeal cyst Laryngitis Laryngopharyngeal reflux (LPR) Laryngospasm vocal cords Laryngopharyngeal reflux (LPR) Vocal fold nodule Vocal fold paresis Vocal cord dysfunction epiglottis Epiglottitis trachea Tracheitis Laryngotracheal stenosis Lower RT / lung disease (including LRTIs ) Bronchial / obstructive acute Acute bronchitis chronic COPD Chronic bronchitis Acute exacerbation of COPD ) Asthma ( Status asthmaticus Aspirin-induced Exercise-induced Bronchiectasis Cystic fibrosis unspecified Bronchitis Bronchiolitis Bronchiolitis obliterans Diffuse panbronchiolitis Interstitial / restrictive ( fibrosis ) External agents/ occupational lung disease Pneumoconiosis Aluminosis Asbestosis Baritosis Bauxite fibrosis Berylliosis Caplan's syndrome Chalicosis Coalworker's pneumoconiosis Siderosis Silicosis Talcosis Byssinosis Hypersensitivity pneumonitis Bagassosis Bird fancier's lung Farmer's lung Lycoperdonosis Other ARDS Combined pulmonary fibrosis and emphysema Pulmonary edema Löffler's syndrome / Eosinophilic pneumonia Respiratory hypersensitivity Allergic bronchopulmonary aspergillosis Hamman-Rich syndrome Idiopathic pulmonary fibrosis Sarcoidosis Vaping-associated pulmonary injury Obstructive / Restrictive Pneumonia / pneumonitis By pathogen Viral Bacterial Pneumococcal Klebsiella Atypical bacterial Mycoplasma Legionnaires' disease Chlamydiae Fungal Pneumocystis Parasitic noninfectious Chemical / Mendelson's syndrome Aspiration / Lipid By vector/route Community-acquired Healthcare-associated Hospital-acquired By distribution Broncho- Lobar IIP UIP DIP BOOP-COP NSIP RB Other Atelectasis circulatory Pulmonary hypertension Pulmonary embolism Lung abscess Pleural cavity / mediastinum Pleural disease Pleuritis/pleurisy Pneumothorax / Hemopneumothorax Pleural effusion Hemothorax Hydrothorax Chylothorax Empyema/pyothorax Malignant Fibrothorax Mediastinal disease Mediastinitis Mediastinal emphysema Other/general Respiratory failure Influenza Common cold SARS Coronavirus disease 2019 Idiopathic pulmonary haemosiderosis Pulmonary alveolar proteinosis v t e Diseases of joints General Arthritis Monoarthritis Oligoarthritis Polyarthritis Symptoms Joint pain Joint stiffness Inflammatory Infectious Septic arthritis Tuberculosis arthritis Crystal Chondrocalcinosis CPPD (Psudogout) Gout Seronegative Reactive arthritis Psoriatic arthritis Ankylosing spondylitis Other Juvenile idiopathic arthritis Rheumatoid arthritis Felty's syndrome Palindromic rheumatism Adult-onset Still's disease Noninflammatory Hemarthrosis Osteoarthritis Heberden's node Bouchard's nodes Osteophyte
-
Bacterial Pneumonia
Wikipedia
External links [ edit ] Classification D ICD - 10 : J13 - J16 ICD - 9-CM : 481 - 483 MeSH : D018410 External resources eMedicine : emerg/465 v t e Diseases of the respiratory system Upper RT (including URTIs , common cold ) Head sinuses Sinusitis nose Rhinitis Vasomotor rhinitis Atrophic rhinitis Hay fever Nasal polyp Rhinorrhea nasal septum Nasal septum deviation Nasal septum perforation Nasal septal hematoma tonsil Tonsillitis Adenoid hypertrophy Peritonsillar abscess Neck pharynx Pharyngitis Strep throat Laryngopharyngeal reflux (LPR) Retropharyngeal abscess larynx Croup Laryngomalacia Laryngeal cyst Laryngitis Laryngopharyngeal reflux (LPR) Laryngospasm vocal cords Laryngopharyngeal reflux (LPR) Vocal fold nodule Vocal fold paresis Vocal cord dysfunction epiglottis Epiglottitis trachea Tracheitis Laryngotracheal stenosis Lower RT / lung disease (including LRTIs ) Bronchial / obstructive acute Acute bronchitis chronic COPD Chronic bronchitis Acute exacerbation of COPD ) Asthma ( Status asthmaticus Aspirin-induced Exercise-induced Bronchiectasis Cystic fibrosis unspecified Bronchitis Bronchiolitis Bronchiolitis obliterans Diffuse panbronchiolitis Interstitial / restrictive ( fibrosis ) External agents/ occupational lung disease Pneumoconiosis Aluminosis Asbestosis Baritosis Bauxite fibrosis Berylliosis Caplan's syndrome Chalicosis Coalworker's pneumoconiosis Siderosis Silicosis Talcosis Byssinosis Hypersensitivity pneumonitis Bagassosis Bird fancier's lung Farmer's lung Lycoperdonosis Other ARDS Combined pulmonary fibrosis and emphysema Pulmonary edema Löffler's syndrome / Eosinophilic pneumonia Respiratory hypersensitivity Allergic bronchopulmonary aspergillosis Hamman-Rich syndrome Idiopathic pulmonary fibrosis Sarcoidosis Vaping-associated pulmonary injury Obstructive / Restrictive Pneumonia / pneumonitis By pathogen Viral Bacterial Pneumococcal Klebsiella Atypical bacterial Mycoplasma Legionnaires' disease Chlamydiae Fungal Pneumocystis Parasitic noninfectious Chemical / Mendelson's syndrome Aspiration / Lipid By vector/route Community-acquired Healthcare-associated Hospital-acquired By distribution Broncho- Lobar IIP UIP DIP BOOP-COP NSIP RB Other Atelectasis circulatory Pulmonary hypertension Pulmonary embolism Lung abscess Pleural cavity / mediastinum Pleural disease Pleuritis/pleurisy Pneumothorax / Hemopneumothorax Pleural effusion Hemothorax Hydrothorax Chylothorax Empyema/pyothorax Malignant Fibrothorax Mediastinal disease Mediastinitis Mediastinal emphysema Other/general Respiratory failure Influenza Common cold SARS Coronavirus disease 2019 Idiopathic pulmonary haemosiderosis Pulmonary alveolar proteinosis v t e Pneumonia Infectious pneumonias Bacterial pneumonia Viral pneumonia Fungal pneumonia Parasitic pneumonia Atypical pneumonia Community-acquired pneumonia Healthcare-associated pneumonia Hospital-acquired pneumonia Ventilator-associated pneumonia Severe acute respiratory syndrome Pneumonias caused by infectious or noninfectious agents Aspiration pneumonia Lipid pneumonia Eosinophilic pneumonia Bronchiolitis obliterans organizing pneumonia Noninfectious pneumonia Chemical pneumonitis Idiopathic pneumonia syndromeSFTPB, CXCL1, F2, CYP2J2, CXCL2, PDPN, ITGB3, PECAM1, TLR2, TLR6, SFTPA1, SFTPC, SFTPD, CAT, IL6, IL22, IL10, HMGB1, HAMP, IL17A, NAMPT, IP6K1, IL17RA, SCGB1A1, TYK2, TNF, ABL1, FOXP3, IL23A, IL36G, ACE2, BPIFB1, UCN3, NLRP3, MIR155, MIR21, CCR2, DEFB4B, LARP1BP2, TLR3, SERPINA1, STAT3, GRN, CD68, CHI3L1, CR1, CRP, CSF2, CSF3, CST3, DEFB4A, DNAH8, FER, NR3C1, CCL2, HIF1A, HPR, IL11, LCN2, MTTP, NM, SERPINE1, PF4, AMBP, S100A12, RNU6-392P
-
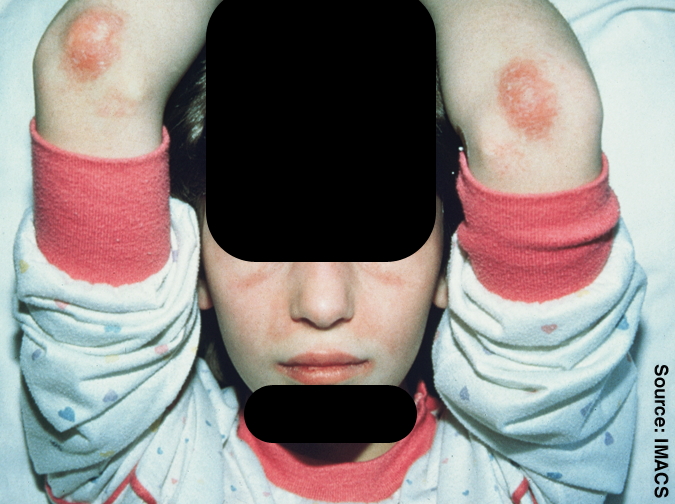
Juvenile Dermatomyositis
Wikipedia
Please help to improve this article by introducing more precise citations. ( April 2009 ) ( Learn how and when to remove this template message ) Juvenile dermatomyositis Juvenile dermatomyositis Specialty Rheumatology Juvenile dermatomyositis ( JDM ) is an idiopathic inflammatory myopathy (IMM) of presumed autoimmune dysfunction resulting in muscle weakness among other complications.TNF, IL1B, IL1A, C2, C9, HLA-B, HLA-DQA1, HLA-DRB1, RBM45, VCAM1, CCL21, IFNA13, IFNA1, MX1, TLR7, IFIH1, TLR3, THBS1, CDR3, TLR4, CRISP2, ABCC8, TRIM21, WT1, ACR, CD83, ISG15, MORC3, IGAN1, SMOC1, MCHR2, NT5C1A, SPZ1, ABCC11, PRSS55, SPP1, PLCL1, CCL19, IFNG, BLK, C4A, CASP6, CD68, CRP, HLA-A, HLA-DMA, HLA-DMB, IFIT3, IFNB1, IL6, CCL18, CXCL8, IL10, CXCL10, IRF7, LGALS9, NCAM1, PF4, BCL2, PLCG2, S100A8, MIR10A