PMID 9631643 . ^ Owen MJ, Norcross-Nechay K, Howie VM (January 1993). "Brainstem auditory evoked potentials in young children before and after tympanostomy tube placement". ... PMID 8436453 . ^ Eric Lupo J, Koka K, Thornton JL, Tollin DJ (February 2011).
This risk is greater in males than females. [31] See also [ edit ] Shoulder problems References [ edit ] ^ a b c d e f g h i j k l m n o Bonz, J; Tinloy, B (May 2015). ... PMID 24939380 . ^ Wasserstein, DN; Sheth, U; Colbenson, K; Henry, PD; Chahal, J; Dwyer, T; Kuhn, JE (December 2016).
Overview A dislocated shoulder is an injury in which the upper arm bone pops out of the cup-shaped socket that's part of the shoulder blade. The shoulder is the body's most flexible joint, which makes it more likely to dislocate. If you suspect a dislocated shoulder, seek prompt medical attention. Most people regain full use of their shoulder within a few weeks. However, once a shoulder dislocates, the joint might be prone to repeat dislocations. Symptoms Dislocated shoulder symptoms can include: A visibly deformed or out-of-place shoulder Swelling or bruising Intense pain Inability to move the joint Shoulder dislocation can also cause numbness, weakness or tingling near the injury, such as in the neck or down the arm.
. ^ Laigle-Donadey F, Benouaich-Amiel A, Hoang-Xuan K, Sanson M (2005). "[Molecular biology of oligodendroglial tumors]". ... Neuro-Oncology . 4 (4): 308–381. doi : 10.1215/15228517-4-4-308 . ^ Anderson K. and Lindsey W., "What is the optimal length of treatment with Temodar (temozolomide) for glioblastoma?"
A rare glial tumor characterized by a highly cellular lesion that is diffusly infiltrating at the periphery and consists of evenly-spaced monomorphic cells with the oligodendroglial phenotype. It typically occurs in the supratentorial white matter. Histologically, the cells are uniformly round to oval with round nuclei, delicate chromatin and small nucleoli. Most patients present with seizures.
Oligodendrogliomas are cerebral tumors that are differentiated from other gliomas on the basis of their unique genetic characteristics and better response to chemotherapy. These tumors are classified according to their grade (low grade oligodendrogliomas: grade II of the WHO classification and anaplastic oligodendrogliomas: grade III of the WHO classification) and according to their pure or mixed histology (oligoastrocytomas). Epidemiology Until now, the incidence of these tumors has been largely underestimated. Oligodendrogliomas may represent up to 30% of all adult gliomas. Annual incidence can therefore be estimated at around one new case per 100,000 individuals per year. Prevalence is estimated at 1/300,000. Clinical description Low grade tumors are usually diagnosed after a prolonged history of seizures and headaches.
Oligodendrogliomas are brain tumors arising from oligodendrocytes, a type of cell that makes up the supportive (glial) tissue of the brain. They can be low-grade (grade II) or high-grade (grade III, also called anaplastic). While they can be found anywhere within the cerebral hemisphere, they are most common in the frontal and temporal lobes. They are generally soft, grayish-pink tumors that often contain mineral deposits (calcifications), areas of hemorrhage, and/or cysts. They tend to grow slowly and may be present for many years before they are diagnosed.
About 16 million cases occur a year, which result in about 25,000 deaths worldwide. [17] References [ edit ] ^ a b c d e f g h i j k l m n o p q r s t u v w x y Anna E. ... PMID 25530442 . ^ Williams, V; Lakshmikantha, KM; Nallasamy, K; Sudeep, KC; Baranwal, AK; Jayashree, M (November 2018).
A rare form of salmonellosis caused by Salmonella enterica serovar Paratyphi A, characterized by typical symptoms of enteric fever including high fever, headache, abdominal pain and intestinal symptoms, dry cough, chills, and rashes, followed by a long period of recovery. The infection can be complicated by intestinal hemorrhage and perforation, as well as cardiac involvement, and may even be fatal. Transmission of the pathogen is via the fecal-oral route, with humans as the sole reservoir of infection.
.; Ferber, Reed; Wiley, J. Preston; Boyd, Steven K. (Fall 2011). "Bone Quality and Muscle Strength in Female Athletes with Lower Limb Stress Fractures". ... "Theropod stress fractures and tendon avulsions as a clue to activity", in Mesozoic Vertebrate Life , edited by Tanke, D. H., and Carpenter, K., Indiana University Press, p. 331–336.
Overview Stress fractures are tiny cracks in a bone. They're caused by repetitive force, often from overuse — such as repeatedly jumping up and down or running long distances. Stress fractures can also develop from normal use of a bone that's weakened by a condition such as osteoporosis. Stress fractures are most common in the weight-bearing bones of the lower leg and foot. Track and field athletes and military recruits who carry heavy packs over long distances are at highest risk, but anyone can sustain a stress fracture. If you start a new exercise program, for example, you might develop stress fractures if you do too much too soon.
March fracture Other names Fatigue fracture or Stress fracture of metatarsal bone Stress fracture of the second metatarsal bone Specialty Orthopedic March fracture , is the fracture of the distal third of one of the metatarsals occurring because of recurrent stress. It is more common in soldiers, but also occurs in hikers, organists, people whose duties entail much standing (such as hospital doctors). March fractures most commonly occur in the second and third metatarsal bones of the foot. [1] [2] [3] It is a common cause of foot pain, especially when people suddenly increase their activities. [4] Contents 1 Signs and symptoms 2 Diagnosis 2.1 Differential diagnosis 3 Treatment 4 Occurrence 5 References 6 External links Signs and symptoms [ edit ] The onset is not dramatic. When the boot or shoes are taken off, there is a cramp-like pain in the affected forefoot, and moderate local edema appears on the dorsal aspect. On moving each toe in turn, that of the involved metatarsal causes pain, and when the bone is palpated from the dorsal surface, a point of tenderness is found directly over the lesion.
Definitive options include: [8] Bonded intra-coronal restoration Onlay restoration, either direct or indirectly placed (currently the recommended technique) Crown restoration (though this is associated with a high incidence of loss of vitality in teeth with CTS) Teeth originally presenting with CTS may subsequently require Root Canal therapy (if pain persists after above) or Extraction History [ edit ] The term "cuspal fracture odontalgia" was suggested in 1954 by Gibbs. [1] Subsequently, the term "cracked tooth syndrome" was coined in 1964 by Cameron, [2] who defined the condition as "an incomplete fracture of a vital posterior tooth that involves the dentin and occasionally extends into the pulp." [1] References [ edit ] ^ a b c d e f g h i j k l m n o p q r s Banerji, S; Mehta, SB; Millar, BJ (May 22, 2010). ... PMID 20489766 . ^ a b c d e f g Mathew, S; Thangavel, B; Mathew, CA; Kailasam, S; Kumaravadivel, K; Das, A (Aug 2012). "Diagnosis of cracked tooth syndrome" .
Niemann–Pick disease Pronunciation / n iː m ən ˈ p ɪ k / nee-mən- PIK ) [1] Specialty Medical genetics Niemann–Pick disease is a group of severe inherited metabolic disorders , in which sphingomyelin accumulates in lysosomes in cells (the lysosomes normally transport material through and out of cells). ... PMID 11787939 . ^ Liu, B.; Turley, S. D.; Burns, D. K.; Miller, A. M.; Repa, J. J.; Dietschy, J.
Niemann-Pick disease (NPD) is a group of inherited metabolic disorders in which harmful quantities of a fatty substance (lipids) accumulate in the spleen, liver, lungs, bone marrow, and brain. Symptoms may include lack of muscle coordination, brain degeneration, learning problems, loss of muscle tone, increased sensitivity to touch, spasticity, feeding and swallowing difficulties, slurred speech, and an enlarged liver and spleen. Inheritance is autosomal recessive. Niemann-Pick disease is divided into four main types according to the altered (mutated) gene and the signs and symptoms: Type A , caused by mutations in the SMPD1 gene. It is the most severe form, occurs in early infancy and is seen primarily in Jewish families. Type B , caused by mutations in the SMPD1 gene. Usually occurs in children, and affects the liver, spleen and lungs (visceral form), but generally does not affect the brain.
Overview Niemann-Pick is a rare, inherited disease that affects the body's ability to metabolize fat (cholesterol and lipids) within cells. These cells malfunction and, over time, die. Niemann-Pick disease can affect the brain, nerves, liver, spleen, bone marrow and, in severe cases, lungs. People with this condition experience symptoms related to progressive loss of function of nerves, the brain and other organs. Niemann-Pick can occur at any age but mainly affects children. The disease has no known cure and is sometimes fatal. Treatment is focused on helping people live with their symptoms. Niemann-Pick care at Mayo Clinic Symptoms Niemann-Pick signs and symptoms may include: Clumsiness and difficulty walking Excessive muscle contractions (dystonia) or eye movements Sleep disturbances Difficulty swallowing and eating Recurrent pneumonia The three main types of Niemann-Pick are types A, B and C.
PMID 17020422 . ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al am an ao ap James, William; Berger, Timothy; Elston, Dirk (2005). ... CS1 maint: extra text: authors list ( link ) ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah author., Marks, James G., Jr. (2019).
A form of cutaneous lupus erythematosus (CLE) that includes five different forms: discoid lupus erythematosus (DLE), chilblain lupus, hypertrophic or verrucous lupus erythematosus, lupus erythematosus tumidus, and lupus erythematosus panniculitis.
A rare form of chronic cutaneous lupus erythematosus characterized by erythematous, scaly papules and plaques preferentially occurring on sun-exposed skin areas (scalp, face, and ears) and exhibiting follicular plugging, pigmentary changes, and central atrophy, scarring, and telangiectasia. Skin biopsy shows a perivascular and periadnexal lymphocytic infiltrate and involvement of the dermoepidermal junction with thickening of the basement membrane and vacuolar degeneration of the basal cells. A small percentage of patients may develop systemic lupus erythematosus.
PMC 1876942 . PMID 16682385 . ^ Duckitt K, Harrington D (March 2005). "Risk factors for pre-eclampsia at antenatal booking: systematic review of controlled studies" . ... PMID 17007686 . ^ Olofsson P, Laurini RN, Marsál K (May 1993). "A high uterine artery pulsatility index reflects a defective development of placental bed spiral arteries in pregnancies complicated by hypertension and fetal growth retardation".
Retrieved 12 December 2017 . ^ a b c d e f g h i j k l "Growth Hormone Deficiency" . NORD (National Organization for Rare Disorders) . 2016 . ... PMID 11889216 . ^ Saborio P, Hahn S, Hisano S, Latta K, Scheinman JI, Chan JC (October 1998).
A number sign (#) is used with this entry because of evidence that isolated growth hormone deficiency type III with agammaglobulinemia (IGHD3) is caused by mutation in the BTK gene (300300) on Xq22. Description IGHD3 is characterized by agammaglobulinemia and markedly reduced numbers of B cells, short stature, delayed bone age, and good response to treatment with growth hormone (summary by Conley et al., 1991). For general phenotypic information and a discussion of genetic heterogeneity of IGHD, see 262400. Clinical Features Fleisher et al. (1980) described a kindred in which 2 brothers and 2 sons of their oldest sister had hypogammaglobulinemia deficiency. Recurrent sinopulmonary infections were a prominent feature in 2 patients.
A number sign (#) is used with this entry because of evidence that Kowarski syndrome is caused by mutation in the growth hormone gene (GH1; 139250) on chromosome 17q. Mutation in the GH1 gene also causes several types of isolated growth hormone deficiency (IGHD); see 262400 for a summary. Description Kowarski syndrome, or short stature associated with bioinactive growth hormone, is characterized clinically by normal or slightly increased GH secretion, pathologically low IGF1 (147440) levels, and normal catch-up growth on GH replacement therapy (Besson et al., 2005). Clinical Features Kowarski et al. (1978) studied 2 unrelated 3-year-old boys with growth retardation and delayed bone ages, and with normal immunoreactive growth hormone after stimulation but low levels of somatomedin. Unlike the result in patients with Laron dwarfism (262500), exogenous human growth hormone induced normal levels of somatomedin and a significant increase in growth rate.
A number sign (#) is used with this entry because isolated growth hormone deficiency type IA (IGHD1A) is caused by homozygous or compound heterozygous mutation in the GH1 (139250) on chromosome 17q23. Description Isolated growth hormone deficiency type IA is an autosomal recessive disorder characterized by severe growth failure (SDS less than -4.5) by 6 months of age, undetectable growth hormone (GH) concentrations, and a tendency to develop antibodies despite an initial good response to rhGH treatment (summary by Alatzoglou et al., 2014). Genetic Heterogeneity of Isolated Growth Hormone Deficiency See IGHD1B (617281) and IGHD2 (173100), both caused by mutation in the GH1 gene; IGHD3 (307200), caused by mutation in the BTK gene (300300); IGHD4 (618157), caused by mutation in the GHRHR gene (139191); and IGHD5 (618160), caused by mutation in the RNPC3 gene (618016). Nomenclature In an early classification of IGHD (Phillips and Cogan, 1994), 4 forms of IGHD were based on ihheritance pattern. IGHD IA and IB (612781) were both inherited in an autosomal recessive manner.
Isolated growth hormone deficiency is a condition caused by a severe shortage or absence of growth hormone without other hormonal problems. Growth hormone is a protein necessary for normal growth of the bone and body tissues. Because people with this condition don't have enough of this hormone, they have short stature, which is noticeable from early childhood. There are basically four different types of isolated growth hormone deficiency, which are classified by the severity of the symptoms, the cause and the inheritance: isolated growth hormone deficiency type IA , isolated growth hormone deficiency type IB , isolated growth hormone deficiency type II and isolated growth hormone deficiency type III . Treatment involves giving growth hormone to those who are affected.
Intention tremor became known as part of Charcot's triad [ citation needed ] (not to be confused with the Charcot triad of acute cholangitis) which, along with nystagmus and scanning speech, act as strong indications of MS. [23] References [ edit ] ^ a b c d e f g h i j k l [1] Archived 2011-07-21 at the Wayback Machine Seeberger, Lauren. ... PMID 16344298 . ^ Deuschl, G.; Wenzelburger, R; Löffler, K; Raethjen, J; Stolze, H (2000). "Essential tremor and cerebellar dysfunction Clinical and kinematic analysis of intention tremor" .
Other clinical signs, such as hirsutism, potbellied appearance, muscle wasting, laminitic episodes, and increased predisposition to infection usually take between 30 days and 1 year to improve. [11] Cyproheptadine may be added to the treatment regime in horses that are inadequately responding to pergolide, [11] but is usually only used in horses with advanced PPID on high doses of pergolide. [13] See also [ edit ] Equine metabolic syndrome Henneke horse body condition scoring system References [ edit ] ^ a b c d e f g h i j k l m n o p q r s Stephen M. Reed; Warwick M. ... Equine Veterinary Journal, 45: 74–79. doi: 10.1111/j.2042-3306.2012.00578 ^ a b c d e f g Durham, A. E., McGowan, C. M., Fey, K., Tamzali, Y. and van der Kolk, J.
J Shoulder Elbow Surg . 12 : 314–21. doi : 10.1016/s1058-2746(03)00030-2 . ^ Sarkar, K; Taine, W; Uhthoff, HK (1990). "The ultrastructure of the coracoacromial ligament in patients with chronic impingement syndrome". ... PMID 3417708 . ^ Pedowitz RA, Yamaguchi K, Ahmad CS, et al. (2012). "American Academy of Orthopaedic Surgeons Clinical Practice Guideline on optimizing the management of rotator cuff problems".
. ^ Kuźma, E. B.; Llewellyn, D. J.; Langa, K. M.; Wallace, R. B.; Lang, I. A. (2014). ... A.; Eaves, D. W.; Smith, A. R.; Nixon, K. (2009). "Alcohol inhibition of neurogenesis: A mechanism of hippocampal neurodegeneration in an adolescent alcohol abuse model" .
.; Fennell, E. B.; Gilmore, R. L.; Heilman, K. M. (1995). "Dissociation of anosognosia for hemiplegia and aphasia during left-hemisphere anesthesia". ... S2CID 46383489 . INIST : 3452304 . ^ Heilman, K. M.; Barrett, A. M.; Adair, J. C. (1998).
Guttmacher Institute . 2007-11-12 . Retrieved 2019-05-22 . ^ a b Lai, K. K. Rebecca (2019-05-15). "Abortion Bans: 8 States Have Passed Bills to Limit the Procedure This Year" .
The children included three siblings and their first cousin; the family was known to be highly consanguineous . [6] [15] References [ edit ] ^ a b Online Mendelian Inheritance in Man (OMIM): Malpuech facial clefting syndrome - 248340 ^ a b Adeleye, A. O.; Olowookere, K. G. (Sep–Oct 2010). "A Human Tail in an Infant (Letter)". ... PMID 15793834 . ^ a b c Chinen, Y.; Naritomi, K. (Dec 1995). "Malpuech facial clefting syndrome in a Japanese boy with cardiac defects" .
A number sign (#) is used with this entry because of evidence that 3MC syndrome-3 (3MC3) is caused by compound heterozygous mutation in the COLEC10 gene (607620) on chromosome 8q24. Description The term '3MC syndrome' encompasses 4 rare autosomal recessive disorders that were previously designated the Carnevale, Mingarelli, Malpuech, and Michels syndromes, respectively. The main features of these syndromes are facial dysmorphism that includes hypertelorism, blepharophimosis, blepharoptosis, and highly arched eyebrows, which are present in 70 to 95% of cases. Cleft lip and palate, postnatal growth deficiency, cognitive impairment, and hearing loss are also consistent findings, occurring in 40 to 68% of cases. Craniosynostosis, radioulnar synostosis, and genital and vesicorenal anomalies occur in 20 to 30% of cases.
3MC syndrome describes a rare developmental disorder, that unifies the overlapping autosomal recessive disorders previously known as Carnevale, Mingarelli, Malpuech and Michels syndromes, characterized by a spectrum of developmental anomalies that include distinctive facial dysmorphism (i.e. hypertelorism, blepharophimosis, blepharoptosis, highly arched eyebrows), cleft lip and/or palate, craniosynostosis, learning disability, radioulnar synostosis and genital and vesicorenal anomalies. Less common features reported include anterior chamber defects, cardiac anomalies (e.g. ventricular septal defect; see this term), caudal appendage, umbilical hernia/omphalocele and diastasis recti.
Neuropsychologia, 41(10), 1290-1295. doi : 10.1016/s0028-3932(03)00062-9 . ^ a b c d e f Meador, K. J., Allen, M. E., Adams, R. J., & Loring, D. ... Brain. 1882;4:153-168. ^ http://medical.yourdictionary.com/symmetrical-gangrene ^ a b c d e f g h i j k l m n Jones E. The precise diagnostic value of allochiria.
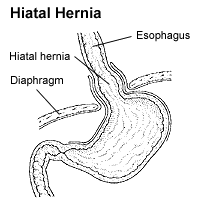
With proper patient selection, laparoscopic fundoplication recent studies have indicated relatively low complication rates, quick recovery, and relatively good long term results. [14] [15] [16] [17] [18] Epidemiology [ edit ] Incidence of hiatal hernias increases with age; approximately 60% of individuals aged 50 or older have a hiatal hernia. [19] Of these, 9% are symptomatic, depending on the competence of the lower esophageal sphincter (LES). 95% of these are "sliding" hiatal hernias, in which the LES protrudes above the diaphragm along with the stomach, and only 5% are the "rolling" type (paraesophageal), in which the LES remains stationary, but the stomach protrudes above the diaphragm. [ citation needed ] Hiatal hernias are most common in North America and Western Europe and rare in rural African communities. [20] Some have proposed that insufficient dietary fiber and the use of a high sitting position for defecation may increase the risk. [21] References [ edit ] ^ a b c d e f g h i j k l m n o p q r s t u v w Roman, S; Kahrilas, PJ (23 October 2014). ... Archived (PDF) from the original on 27 February 2015. ^ Goyal Raj K, "Chapter 286. Diseases of the Esophagus".
Overview A hiatal hernia occurs when the upper part of your stomach bulges through the large muscle separating your abdomen and chest (diaphragm). Your diaphragm has a small opening (hiatus) through which your food tube (esophagus) passes before connecting to your stomach. In a hiatal hernia, the stomach pushes up through that opening and into your chest. A small hiatal hernia usually doesn't cause problems. You may never know you have one unless your doctor discovers it when checking for another condition. But a large hiatal hernia can allow food and acid to back up into your esophagus, leading to heartburn.
Goodman et al. (1969) observed 6 affected persons in 2 generations. Five of the 6 were female. This disorder is sometimes called congenital short esophagus (Myles, 1939) or partial thoracic stomach. Carre and Froggatt (1970) described 8 definite cases in 3 successive generations of a family. Others were equivocally affected. Sidd et al. (1966) observed a sliding hiatal hernia in 4 brothers, aged 42 to 44 years, 2 of whom were monozygotic twins. Carre et al. (1999) described a 5-generation family in which 23 of 38 individuals had a radiologically proven hiatus hernia.