-
Progressive External Ophthalmoplegia With Mitochondrial Dna Deletions, Autosomal Recessive 4
OMIM
Clinical Features Ronchi et al. (2012) reported 6 patients, including 2 sibs, with adult-onset mitochondrial myopathy. Three patients and the 2 sibs presented between 40 and 69 years of age with slowly progressive external ophthalmoplegia (in 2 patients) and/or limb muscle weakness. ... She was then well until age 20 years, when she experienced 2 episodes of acute mitochondrial myopathy with proximal muscle weakness, increased serum creatine kinase, and rhabdomyolysis possibly triggered by infection. ... INHERITANCE - Autosomal recessive HEAD & NECK Ears - Sensorineural deafness (family A) Eyes - Progressive external ophthalmoplegia (in most patients) - Ptosis (in most patients) ABDOMEN Gastrointestinal - Dysphagia (in some patients) MUSCLE, SOFT TISSUES - Mitochondrial myopathy - Muscle weakness, proximal - Weakness primarily affects lower limbs - Upper limbs may show muscle weakness - Muscle weakness, distal (family A) - Muscle atrophy - Muscle biopsy shows fiber size variability - Ragged-red fibers - COX-negative fibers - MtDNA deletions - Myopathic changes seen on EMG NEUROLOGIC Central Nervous System - Cognitive impairment (family A) - Cortical atrophy (family A) Peripheral Nervous System - Axonal neuropathy (family A) - Hyporeflexia (family A) VOICE - Dysphonia LABORATORY ABNORMALITIES - Increased serum creatine kinase, mild - Increased serum lactate, mild MISCELLANEOUS - Onset in adulthood - Variable presentation and phenotype MOLECULAR BASIS - Caused by mutation in the deoxyguanosine kinase gene (DGUOK, 601465.0008 ) ▲ Close
-
Hypotonia
Wikipedia
Phelan–McDermid syndrome 3-Methylcrotonyl-CoA carboxylase deficiency [4] Achondroplasia Aicardi syndrome Autism spectrum disorders [5] Canavan disease Centronuclear myopathy (including myotubular myopathy) Central core disease CHARGE syndrome Cohen syndrome Costello syndrome Dejerine–Sottas disease (HMSN Type III) Down syndrome a.k.a. trisomy 21 — most common Ehlers–Danlos syndrome Familial dysautonomia (Riley–Day syndrome) FG syndrome Fragile X syndrome Griscelli syndrome Type 1 (Elejalde syndrome) Disorder Growth Hormone Disorder Pituitary Dwarfism Holocarboxylase synthetase deficiency / Multiple carboxylase deficiency [6] Krabbe disease Leigh's disease Lesch–Nyhan syndrome [7] Marfan's syndrome Menkes syndrome Methylmalonic acidemia Myotonic dystrophy Niemann–Pick disease Nonketotic hyperglycinemia (NKH) or Glycine encephalopathy (GCE) Noonan syndrome Neurofibromatosis Patau syndrome a.k.a. trisomy 13 Prader–Willi syndrome Rett syndrome Septo-optic dysplasia (de Morsier syndrome) Snyder–Robinson syndrome (SRS) Spinal muscular atrophy (SMA) Succinic semialdehyde dehydrogenase deficiency (SSADH) Tay–Sachs disease Werdnig–Hoffmann syndrome – Spinal muscular atrophy with congenital degeneration of anterior horns of spinal cord. ... External links [ edit ] hypotonia at NINDS Hypotonia – Medline Plus Classification D ICD - 10 : P94.2 ICD - 9-CM : 358 , 781.3 MeSH : D009123 DiseasesDB : 21417 External resources MedlinePlus : 003298 v t e Diseases of muscle , neuromuscular junction , and neuromuscular disease Neuromuscular- junction disease autoimmune Myasthenia gravis Lambert–Eaton myasthenic syndrome Neuromyotonia Myopathy Muscular dystrophy ( DAPC ) AD Limb-girdle muscular dystrophy 1 Oculopharyngeal Facioscapulohumeral Myotonic Distal (most) AR Calpainopathy Limb-girdle muscular dystrophy 2 Congenital Fukuyama Ullrich Walker–Warburg XR dystrophin Becker's Duchenne Emery–Dreifuss Other structural collagen disease Bethlem myopathy PTP disease X-linked MTM adaptor protein disease BIN1-linked centronuclear myopathy cytoskeleton disease Nemaline myopathy Zaspopathy Channelopathy Myotonia Myotonia congenita Thomsen disease Neuromyotonia / Isaacs syndrome Paramyotonia congenita Periodic paralysis Hypokalemic Thyrotoxic Hyperkalemic Other Central core disease Mitochondrial myopathy MELAS MERRF KSS PEO General Inflammatory myopathy Congenital myopathy v t e Conditions originating in the perinatal period / fetal disease Maternal factors complicating pregnancy, labour or delivery placenta Placenta praevia Placental insufficiency Twin-to-twin transfusion syndrome chorion / amnion Chorioamnionitis umbilical cord Umbilical cord prolapse Nuchal cord Single umbilical artery presentation Breech birth Asynclitism Shoulder presentation Growth Small for gestational age / Large for gestational age Preterm birth / Postterm pregnancy Intrauterine growth restriction Birth trauma scalp Cephalohematoma Chignon Caput succedaneum Subgaleal hemorrhage Brachial plexus injury Erb's palsy Klumpke paralysis Affected systems Respiratory Intrauterine hypoxia Infant respiratory distress syndrome Transient tachypnea of the newborn Meconium aspiration syndrome Pleural disease Pneumothorax Pneumomediastinum Wilson–Mikity syndrome Bronchopulmonary dysplasia Cardiovascular Pneumopericardium Persistent fetal circulation Bleeding and hematologic disease Vitamin K deficiency bleeding HDN ABO Anti-Kell Rh c Rh D Rh E Hydrops fetalis Hyperbilirubinemia Kernicterus Neonatal jaundice Velamentous cord insertion Intraventricular hemorrhage Germinal matrix hemorrhage Anemia of prematurity Gastrointestinal Ileus Necrotizing enterocolitis Meconium peritonitis Integument and thermoregulation Erythema toxicum Sclerema neonatorum Nervous system Perinatal asphyxia Periventricular leukomalacia Musculoskeletal Gray baby syndrome muscle tone Congenital hypertonia Congenital hypotonia Infections Vertically transmitted infection Neonatal infection rubella herpes simplex mycoplasma hominis ureaplasma urealyticum Omphalitis Neonatal sepsis Group B streptococcal infection Neonatal conjunctivitis Other Miscarriage Perinatal mortality Stillbirth Infant mortality Neonatal withdrawalNSD2, MSL3, RRM2B, MECP2, LETM1, KANSL1, NUDT2, PPP2CA, PAX7, INTS1, ASCC1, LSS, P4HTM, DDC, CHST14, RALGAPA1, ABAT, SLC25A3, MPZ, CDKL5, PREPL, GLDC, NDUFS7, AP4B1-AS1, SYNE2, VPS13A, KIAA0556, CAMTA1, SATB2, RPGRIP1L, CLASP1, SYNE1, SPECC1L, ADNP, MED13L, KANK1, MLYCD, CRB1, ETHE1, KAT6B, RBFOX2, ATP6V0A2, PIGN, LEMD3, KCNE5, AIPL1, FLRT3, PMPCA, CIC, CIZ1, PHF8, RAI1, ZMYND11, WDR4, SRCAP, PNPLA6, MAGED2, TTN-AS1, TLK2, B4GAT1, WDR45, SRD5A3-AS1, AP4S1, PLPBP, B4GALT7, MRAS, FASTKD2, FAN1, RAB3GAP1, RAB18, CLUAP1, EMC1, ZNF423, IQSEC2, POGZ, PLXND1, RAB3GAP2, NDUFAF3, NIPBL, KMT5B, ALG6, POMT2, PET100, DUOX2, TPRKB, CRPPA, EXOSC3, YARS2, NDUFA13, MLXIPL, SEPSECS, NDUFAF1, ZDHHC9, CTCF, TACO1, TMEM216, TIMMDC1, KCNK9, WAC, MBTPS2, TMEM138, NBAS, LIPT1, KLF13, PTRH2, ATP8A2, GMPPA, GMPPB, ANKRD11, NDUFAF4, SIN3A, SH2B1, PARS2, MMACHC, NSMF, SETBP1, KIFBP, RNU4ATAC, B3GAT3, FBXL4, HIBCH, VPS33B, SACS, SLC17A5, TBL2, RPS6KC1, MLH3, ACAD8, B9D1, BBS9, AHDC1, MMADHC, CCDC22, FLVCR1, SETD2, AP4B1, POMT1, MAP3K20, HESX1, OFD1, PPM1D, PEX3, CNTNAP1, KCNAB2, IKBKG, ELP1, CDK13, PEX11B, SUCLG1, SUCLA2, DPM1, FGF17, NPC2, SYNGAP1, ST3GAL5, EIF2B4, EIF2B3, EIF2B2, EIF2B5, AP1S2, PHOX2B, BAZ1B, SEMA5A, SLC7A7, PIGQ, CUL3, CUL4B, GNPAT, PLA2G6, NELFA, YWHAE, ZIC1, PCGF2, BSND, MOGS, TUBA1A, PAX8, BRPF1, ALDH5A1, KAT6A, LHX3, SHOC2, KMT2D, AAAS, HMGA2, DGCR6, LZTR1, ESS2, USP9X, RBM10, SMC1A, LAGE3, ARID1A, COLQ, TBX19, SMC3, BUB3, KIAA0586, ZEB2, SEC24D, FIG4, WASHC5, AMMECR1, MED12, DGCR2, SCO2, AKT3, FARSB, LRPPRC, AASS, ALG3, CD96, COQ7, HNRNPR, APC2, LAMC3, RXYLT1, SEMA3A, TUBB4A, PIBF1, COG5, ZBTB18, DEAF1, PTDSS1, TMEM94, HACD1, HDAC4, LARGE1, LRAT, ADGRG1, TRIP13, TRIP12, SNAP29, LONP1, COG1, HS6ST1, PEX16, EIF2AK3, CHST3, PIGL, TBX4, ADAMTS2, MPDU1, GTF2IRD1, SEC24C, ZNF592, IQCB1, CEP57, RUBCN, SEMA3E, CEP104, IFT140, INPP5K, BCL11A, HCN1, CEP41, RFT1, NDUFAF2, COG7, MTSS2, GDF6, STRADA, GORAB, TIMM50, PGAP3, TRMT10A, TBCK, FOXP2, PRRT2, EARS2, STX1B, FDX2, TP53RK, NACC1, MYMK, SLC46A1, SLC52A3, TOE1, CCDC151, COX20, ANTXR2, CYP2R1, ATPAF2, PKDCC, TMEM67, UBE3B, PUS1, WNT10A, SLC19A3, ASXL3, SLC2A10, CLPB, TRIM8, SPRY4, LAS1L, TMEM47, TRAPPC9, ADGRV1, POMK, PHF6, COA8, COG8, KISS1R, KDM2B, ORAI1, POMGNT2, WDR73, COX14, USP45, ATCAY, LHX4, MTFMT, METTL23, NUBPL, IYD, TUBB, HYLS1, HEPACAM, JMJD1C, STAC3, EBF3, FEZF1, BRWD3, NALCN, ASPM, MYO1H, CHAMP1, SLC13A5, CANT1, LAMA1, WDR62, CCDC141, DOK7, MMAB, FLG-AS1, SLC6A19, COA6, NEXMIF, RD3, ZFP57, TUBB2B, ARL13B, UBR1, ARID2, ASXL1, SPNS2, WDR81, NDUFA11, B3GALT6, DMBX1, TBC1D20, PROKR2, DIS3L2, NDUFAF6, AMER1, PTCHD1, SLC34A3, A2ML1, RDH12, B3GALNT2, SIK1, CKAP2L, CEP120, VPS13B, FAM120AOS, SPRED1, LGI4, MMAA, LCA5, ARX, WDR26, ARMC9, NANS, ALG1, PACS1, WDR11, NUP133, AGK, NGLY1, MBD5, LMBRD1, SPATA7, TMEM126B, HDAC8, ZC4H2, NDUFA12, INPP5E, PRR12, TWNK, MCCC1, MRPS22, COQ8A, HYMAI, RPGRIP1, C12orf4, NUP107, THOC2, SELENON, MCOLN1, THAP11, PEX26, PIGV, OSGEP, CHD7, RIN2, D2HGDH, RETREG1, RNF216, DGCR8, BDNF-AS, TMCO1, CLIP2, MAGEL2, NR2F1-AS1, DUOXA2, PDP1, IL17RD, AHI1, MKS1, TMEM70, OXSM, RMND1, FKBP14, DARS2, SETD5, ASXL2, SLC35C1, FOXRED1, POMGNT1, TBC1D24, ARID1B, CEP290, TCTN1, TMEM237, UPF3B, WNK1, CPLANE1, ALG12, NDUFAF5, FKRP, GNPTAB, TMEM43, THOC6, SLC52A2, TMEM231, SRD5A3, ARHGAP31, TBL1XR1, BBS10, SLC25A22, ALG9, EHMT1, CAMKMT, CSPP1, TCTN2, ALG13, L2HGDH, CENPT, NAA15, BCL11B, NMNAT1, SIL1, LMBR1, HECW2, PCDH19, HACE1, CC2D2A, ALS2, CHD8, ZSWIM6, PRX, EPG5, WDR19, PRUNE1, KMT2C, TRPV4, SLC25A19, PROK2, TRAPPC11, PIEZO2, ZNF335, PRDM16, MCCC2, IFIH1, IRF2BPL, SEMA4A, STRA6, NSD1, NDUFB11, UFD1, VRK1, GAMT, FLT4, FMR1, MTOR, FUCA1, GAA, GABRA1, GABRB3, GABRD, GABRG2, GALC, GALE, GALNS, GATA1, FLG, GBA, GBE1, GCDH, GCK, GCSH, GDI1, GDNF, B4GALT1, GJA1, GJA8, GJB1, GPC3, FLII, FOXE1, GLI2, ETFB, EIF2B1, EIF2S3, ELN, EMD, EP300, CLN8, ERCC1, ERCC2, ERCC5, ERCC6, ERF, ETFA, ETFDH, FOXG1, EXT1, EZH2, ACSL4, FBN1, FBP1, FKTN, GPC4, FGF8, FGFR1, FGFR3, FH, FHL1, GLE1, GM2A, VLDLR, KCNB1, IDUA, IMPDH1, INS, INPPL1, PDX1, ITGA3, ITGA7, ITGB6, ITPR1, ANOS1, KARS1, KCNA2, KCNC3, HSPD1, KCNH1, KCNJ10, KCNJ11, KCNJ13, KCNQ2, KCNQ3, KIT, KIF11, KIF22, KRAS, LAMA2, LAMB2, IDH2, HRAS, GNAO1, HSD17B10, GNB1, GP1BB, GRID2, GRIN1, GRIN2B, GRM7, MSH6, GTF2I, GUSB, GUCY2D, H1-4, H3-3A, HADHA, HPRT1, HADHB, HARS1, HBA1, HBA2, HBB, HEXA, HLCS, HMGCL, HNRNPH2, HNRNPK, HNRNPU, HPD, EGR2, EFNB1, EEF1A2, BMP2, ATP2B3, ALDH7A1, ATP6V1A, ATP6AP1, ATP7A, ATRX, KIF1A, BBS2, BCKDHA, BCKDHB, BCS1L, BDNF, BMPR1A, RERE, BRAF, BRCA2, BTD, BUB1, BUB1B, CA8, CACNA1A, CACNA1C, CAMK2A, CAMK2B, CASR, CDKN1C, ATP1A3, ATIC, EDNRB, AGA, ACADM, ACADS, ACADSB, ACADVL, ACAT1, ACOX1, ACP2, ACTA1, ACTB, ACY1, ADCY5, ADCY6, AHSG, ASPA, ALAD, ALDH3A2, ALPL, AMT, ANK3, SLC25A4, FAS, AR, ARL3, ARSA, ARVCF, ASCL1, CFL2, CHRNA1, CHRNA7, DNM1, CYP27B1, DAG1, DBH, DBT, DCC, DCX, DDX3X, DES, DGUOK, DHCR7, DLG3, DMD, DMPK, CHRNB2, DNAH5, DYNC1H1, DNMT3A, DOCK3, DPAGT1, DPYD, SLC26A2, DUSP6, DYRK1A, TOR1A, ECHS1, EDN3, CTNND2, CTNNB1, NKX2-5, CSTB, CLCNKA, CLCNKB, CLTC, COL1A1, COL1A2, COL2A1, COL4A1, COL4A2, COL5A1, COL5A2, COL6A2, COL6A3, COL12A1, COMT, COX6B1, COX8A, COX10, COX15, CPS1, CPT1A, CPT2, CRAT, CREBBP, CRX, CRYBA1, LIFR, LIMK1, LMNA, SCN9A, RRAS, RREB1, RYR1, MSMO1, SC5D, ATXN1, ATXN2, SCN1A, SCN1B, SCN2A, SCN4A, SCN8A, SCO1, RPS6KA3, SDHA, SET, SIM1, SIX3, SKI, SLC2A1, SLC3A1, SLC5A5, SLC6A1, SLC6A8, SLC16A2, SLC18A2, RPS20, RPL10, PSAP, RAD21, PTCH1, PTEN, PTPN11, PTS, PURA, PEX19, PEX2, PEX5, PYCR1, ALDH18A1, QDPR, RAC1, RAD51, RPE65, RAF1, RAP1A, RAP1B, RARB, RASA2, RB1, DPF2, RET, REV3L, RFC2, RIT1, RMRP, SLC22A5, SNAI2, SMARCA4, TPO, NR2F1, TG, TGFB2, TGFBR2, THRA, THRB, NKX2-1, TK2, TNXB, TPI1, TPM2, TPM3, TRPS1, SMARCB1, TSHB, TSHR, TTN, TULP1, HIRA, UBA1, UBE2A, UBE3A, ABCB7, SLC35A2, KDM6A, VDR, TCF20, TCF4, TBX1, TACR3, SMARCC2, SMARCE1, SMN1, SMN2, SMPD1, SMS, SON, SOS1, SOS2, SOX2, SOX4, SOX5, SOX9, SOX10, SOX11, SPARC, SPAST, SPG7, SPTBN2, STAT3, STIM1, STXBP1, ABCC8, SURF1, SYT1, LRP5, PSMD12, RELN, NDUFA4, TRNS2, TRNW, TRIM37, MMUT, MVK, MYBPC3, MYL2, MYO5A, NAGA, NDP, NDUFA1, NDUFA2, NDUFA6, TRNQ, NEB, NDUFA9, NDUFA10, NDUFB3, NDUFB9, NDUFB10, NDUFS1, NDUFS2, NDUFS3, NDUFV1, NDUFS4, NDUFS6, TRNS1, TRNN, PRPS1, COX1, LTC4S, EPCAM, MAN2B1, MEF2C, MIPEP, MLH1, KMT2A, MLLT1, MN1, MPI, MSH2, ATP8, COX2, TRNL1, COX3, MTM1, ND1, ND2, ND3, ND4, ND5, ND6, MTTP, MTRR, TRNF, TRNH, NDUFS8, NDUFV2, NEU1, PMP22, PEX10, PEX12, PEX13, PEX14, PHKG2, PHYH, PIK3CA, PLAGL1, PLCB4, PLOD1, PLP1, PMM2, PMS1, NF1, PMS2, POLE, POLG, POU1F1, PPM1B, PPP2R1A, PPP2R5D, PPT1, MAP2K1, MAP2K2, PRODH, PROP1, PEX7, PEX6, PEX1, SLC26A4, NFIX, TONSL, CNOT3, NOTCH1, NOTCH3, PNP, NPC1, NPHP1, NRAS, OCA2, OGDH, OPA1, OPHN1, SIX6, OTX2, P4HB, PAFAH1B1, PRDX1, PAH, PAX6, PC, PCBD1, PCYT1A, PDHA1, EMC1-AS1, ATXN7, ABL1

-
Neuromyotonia
Wikipedia
External links [ edit ] Classification D ICD - 10 : G71.1 ICD - 9-CM : 333.90 MeSH : D020386 DiseasesDB : 31818 External resources Orphanet : 11619 v t e Diseases of muscle , neuromuscular junction , and neuromuscular disease Neuromuscular- junction disease autoimmune Myasthenia gravis Lambert–Eaton myasthenic syndrome Neuromyotonia Myopathy Muscular dystrophy ( DAPC ) AD Limb-girdle muscular dystrophy 1 Oculopharyngeal Facioscapulohumeral Myotonic Distal (most) AR Calpainopathy Limb-girdle muscular dystrophy 2 Congenital Fukuyama Ullrich Walker–Warburg XR dystrophin Becker's Duchenne Emery–Dreifuss Other structural collagen disease Bethlem myopathy PTP disease X-linked MTM adaptor protein disease BIN1-linked centronuclear myopathy cytoskeleton disease Nemaline myopathy Zaspopathy Channelopathy Myotonia Myotonia congenita Thomsen disease Neuromyotonia / Isaacs syndrome Paramyotonia congenita Periodic paralysis Hypokalemic Thyrotoxic Hyperkalemic Other Central core disease Mitochondrial myopathy MELAS MERRF KSS PEO General Inflammatory myopathy Congenital myopathy v t e Diseases of ion channels Calcium channel Voltage-gated CACNA1A Familial hemiplegic migraine 1 Episodic ataxia 2 Spinocerebellar ataxia type-6 CACNA1C Timothy syndrome Brugada syndrome 3 Long QT syndrome 8 CACNA1F Ocular albinism 2 CSNB2A CACNA1S Hypokalemic periodic paralysis 1 Thyrotoxic periodic paralysis 1 CACNB2 Brugada syndrome 4 Ligand gated RYR1 Malignant hyperthermia Central core disease RYR2 CPVT1 ARVD2 Sodium channel Voltage-gated SCN1A Familial hemiplegic migraine 3 GEFS+ 2 Febrile seizure 3A SCN1B Brugada syndrome 6 GEFS+ 1 SCN4A Hypokalemic periodic paralysis 2 Hyperkalemic periodic paralysis Paramyotonia congenita Potassium-aggravated myotonia SCN4B Long QT syndrome 10 SCN5A Brugada syndrome 1 Long QT syndrome 3 SCN9A Erythromelalgia Febrile seizure 3B Paroxysmal extreme pain disorder Congenital insensitivity to pain Constitutively active SCNN1B / SCNN1G Liddle's syndrome SCNN1A / SCNN1B / SCNN1G Pseudohypoaldosteronism 1AR Potassium channel Voltage-gated KCNA1 Episodic ataxia 1 KCNA5 Familial atrial fibrillation 7 KCNC3 Spinocerebellar ataxia type-13 KCNE1 Jervell and Lange-Nielsen syndrome Long QT syndrome 5 KCNE2 Long QT syndrome 6 KCNE3 Brugada syndrome 5 KCNH2 Short QT syndrome KCNQ1 Jervell and Lange-Nielsen syndrome Romano–Ward syndrome Short QT syndrome Long QT syndrome 1 Familial atrial fibrillation 3 KCNQ2 BFNS1 Inward-rectifier KCNJ1 Bartter syndrome 2 KCNJ2 Andersen–Tawil syndrome Long QT syndrome 7 Short QT syndrome KCNJ11 TNDM3 KCNJ18 Thyrotoxic periodic paralysis 2 Chloride channel CFTR Cystic fibrosis Congenital absence of the vas deferens CLCN1 Thomsen disease Myotonia congenita CLCN5 Dent's disease CLCN7 Osteopetrosis A2, B4 BEST1 Vitelliform macular dystrophy CLCNKB Bartter syndrome 3 TRP channel TRPC6 FSGS2 TRPML1 Mucolipidosis type IV Connexin GJA1 Oculodentodigital dysplasia Hallermann–Streiff syndrome Hypoplastic left heart syndrome GJB1 Charcot–Marie–Tooth disease X1 GJB2 Keratitis–ichthyosis–deafness syndrome Ichthyosis hystrix Bart–Pumphrey syndrome Vohwinkel syndrome ) GJB3 / GJB4 Erythrokeratodermia variabilis Progressive symmetric erythrokeratodermia GJB6 Clouston's hidrotic ectodermal dysplasia Porin AQP2 Nephrogenic diabetes insipidus 2 See also: ion channels
-
Ehlers-Danlos Syndrome, Kyphoscoliotic Type, 2
OMIM
Description Ehlers-Danlos syndrome kyphoscoliotic type 2 is characterized by severe muscle hypotonia at birth, progressive scoliosis, joint hypermobility, hyperelastic skin, myopathy, sensorineural hearing impairment, and normal pyridinoline excretion in urine (Baumann et al., 2012). ... Minor criteria include (i) hyperelastic skin with follicular hyperkeratosis, easy bruising, and occasional abnormal scarring; (ii) myopathy as seen clinically by reduced strength and endurance and confirmed in some patients by histology and muscle imaging; and (iii) hearing impairment that is predominantly sensorineural and may not be present in all individuals. Mapping Using genomic DNA from 23 members of an Austrian family segregating autosomal recessive Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss, Baumann et al. (2012) performed a genomewide scan followed by haplotype analysis and autozygosity mapping and identified only 2 linkage regions for which the 2 affected individuals were homozygous identical by descent, on chromosomes 7p15.1 and 16q12.2. ... Molecular Genetics In 2 affected individuals from an Austrian family with Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss, Baumann et al. (2012) sequenced the candidate gene FKBP14 (614505) and identified homozygosity for a 1-bp insertion (614505.0001). ... In a 3-year-old boy with Ehlers-Danlos syndrome and myopathy, Aldeeri et al. (2014) performed exome sequencing and identified homozygosity for a 4-bp splice site deletion in the FKBP14 gene (614505.0003).
-
Chromosome Xp21 Deletion Syndrome
OMIM
Other features included psychomotor retardation, spasticity, growth failure, myopathy, and osteoporosis. Glycerol kinase activity in leukocytes and cultured fibroblasts was less than 5% of controls. ... The infantile form is characterized by adrenal hypoplasia, psychomotor retardation, growth delay, osteoporosis, and in some patients myopathy histologically similar to that of Duchenne muscular dystrophy. ... Patil et al. (1985) also described studies of families in which affected males had combined GK deficiency and adrenal hypoplasia, sometimes with myopathy as well, and had deletion of the Xp21.3-p21.2 region. ... Francke et al. (1987) did mapping studies with various DNA probes in a search for deletions of GK deficiency either in isolation or in association with congenital adrenal hypoplasia and developmental delay with or without congenital dystrophic myopathy. Four of 7 such patients were found to have deletions of different sizes within Xp21. ... In cases with the GKD/adrenal hypoplasia microdeletion syndrome without myopathy, no deletion was found in the dystrophin gene.
-
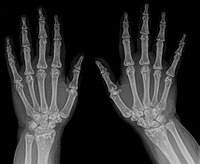
Osteopoikilosis
Wikipedia
External links [ edit ] Classification D ICD - 10 : Q78.8 ICD - 9-CM : 756.53 OMIM : 166700 MeSH : D010023 DiseasesDB : 30071 External resources eMedicine : derm/733 v t e Osteochondrodysplasia Osteodysplasia/ / osteodystrophy Diaphysis Camurati–Engelmann disease Metaphysis Metaphyseal dysplasia Jansen's metaphyseal chondrodysplasia Schmid metaphyseal chondrodysplasia Epiphysis Spondyloepiphyseal dysplasia congenita Multiple epiphyseal dysplasia Otospondylomegaepiphyseal dysplasia Osteosclerosis Raine syndrome Osteopoikilosis Osteopetrosis Other/ungrouped FLNB Boomerang dysplasia Opsismodysplasia Polyostotic fibrous dysplasia McCune–Albright syndrome Chondrodysplasia / chondrodystrophy (including dwarfism ) Osteochondroma osteochondromatosis Hereditary multiple exostoses Chondroma / enchondroma enchondromatosis Ollier disease Maffucci syndrome Growth factor receptor FGFR2 : Antley–Bixler syndrome FGFR3 : Achondroplasia Hypochondroplasia Thanatophoric dysplasia COL2A1 collagen disease Achondrogenesis type 2 Hypochondrogenesis SLC26A2 sulfation defect Achondrogenesis type 1B Autosomal recessive multiple epiphyseal dysplasia Atelosteogenesis, type II Diastrophic dysplasia Chondrodysplasia punctata Rhizomelic chondrodysplasia punctata Conradi–Hünermann syndrome Other dwarfism Fibrochondrogenesis Short rib – polydactyly syndrome Majewski's polydactyly syndrome Léri–Weill dyschondrosteosis v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteins
-
Monilethrix
Wikipedia
External links [ edit ] Classification D ICD - 10 : Q84.1 ( ILDS Q84.140) ICD - 9-CM : 757.4 OMIM : 158000 MeSH : D056734 DiseasesDB : 29592 External resources eMedicine : derm/763 Orphanet : 573 v t e Congenital malformations and deformations of skin appendages Nail disease Anonychia Leukonychia Pachyonychia congenita / Onychauxis Koilonychia Hair disease hypotrichosis /abnormalities: keratin disease Monilethrix IBIDS syndrome Sabinas brittle hair syndrome Pili annulati Pili torti Uncombable hair syndrome Björnstad syndrome Giant axonal neuropathy with curly hair hypertrichosis : Zimmermann–Laband syndrome v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteins This genetic disorder article is a stub .

-
Perimyositis
Wikipedia
Please help improve the article by providing more context for the reader . ( January 2021 ) ( Learn how and when to remove this template message ) Perimyositis Specialty Rheumatology Perimyositis is inflammation or swelling of tissues near the muscles . See also [ edit ] myopathy (muscle disease) myalgia (muscle pain) Masticatory muscle myositis (a disease in dogs) myositis References [ edit ] v t e Inflammation Symptoms Flushing (Rubor) Fever (Calor) Swelling (Tumor) Pain (Dolor) Malaise Mechanism Acute Plasma-derived mediators Bradykinin complement C3 C5a MAC coagulation Factor XII Plasmin Thrombin Cell-derived mediators preformed: Lysosome granules biogenic amines Histamine Serotonin synthesized on demand: cytokines IFN-γ IL-8 TNF-α IL-1 eicosanoids Leukotriene B4 Prostaglandins Nitric oxide Kinins Chronic Macrophage Epithelioid cell Giant cell Granuloma Other Acute-phase reaction Vasodilation Increased vascular permeability Exudate Leukocyte extravasation Chemotaxis Tests Full blood count Leukocytosis C-reactive protein Erythrocyte sedimentation rate General Lymphadenopathy List of inflammed body part states v t e Systemic connective tissue disorders General Systemic lupus erythematosus Drug-induced SLE Libman–Sacks endocarditis Inflammatory myopathy Myositis Dermatopolymyositis Dermatomyositis / Juvenile dermatomyositis Polymyositis * Inclusion body myositis Scleroderma Systemic scleroderma Progressive systemic sclerosis CREST syndrome Overlap syndrome / Mixed connective tissue disease Other hypersensitivity / autoimmune Sjögren syndrome Other Behçet's disease Polymyalgia rheumatica Eosinophilic fasciitis Eosinophilia–myalgia syndrome fibrillin Marfan syndrome Congenital contractural arachnodactyly v t e Symptoms and conditions relating to muscle Pain Myalgia Fibromyalgia Acute Delayed onset Inflammation Myositis Pyomyositis Destruction Muscle weakness Rhabdomyolysis Muscle atrophy / Amyotrophy Other Myositis ossificans Fibrodysplasia ossificans progressiva Compartment syndrome Anterior Diastasis of muscle Diastasis recti Muscle spasm This article about a disease of musculoskeletal and connective tissue is a stub .
-
Becker Muscular Dystrophy
Wikipedia
External links [ edit ] Becker muscular dystrophy at Curlie Classification D ICD - 10 : G71.0 ICD - 9-CM : 359.1 OMIM : 300376 DiseasesDB : 1280 External resources MedlinePlus : 000706 eMedicine : pmr/14 Patient UK : Becker muscular dystrophy Scholia has a topic profile for Becker muscular dystrophy . v t e Muscular dystrophy Types Congenital Dystrophinopathy Becker's Duchenne Distal Emery-Dreifuss Facioscapulohumeral Limb-girdle muscular dystrophy Calpainopathy Myotonic Oculopharyngeal National/International Organizations Muscular Dystrophy Association (USA) Muscular Dystrophy Canada Myotonic Dystrophy Foundation Muskelsvindfonden (Denmark) National/International Events MDA Muscle Walk (USA) Labor Day Telethon (defunct; USA/Canada) Décrypthon (France) Grøn Koncert (Denmark) Clinical trials Stamulumab (MYO-029) Category v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasia v t e Diseases of muscle , neuromuscular junction , and neuromuscular disease Neuromuscular- junction disease autoimmune Myasthenia gravis Lambert–Eaton myasthenic syndrome Neuromyotonia Myopathy Muscular dystrophy ( DAPC ) AD Limb-girdle muscular dystrophy 1 Oculopharyngeal Facioscapulohumeral Myotonic Distal (most) AR Calpainopathy Limb-girdle muscular dystrophy 2 Congenital Fukuyama Ullrich Walker–Warburg XR dystrophin Becker's Duchenne Emery–Dreifuss Other structural collagen disease Bethlem myopathy PTP disease X-linked MTM adaptor protein disease BIN1-linked centronuclear myopathy cytoskeleton disease Nemaline myopathy Zaspopathy Channelopathy Myotonia Myotonia congenita Thomsen disease Neuromyotonia / Isaacs syndrome Paramyotonia congenita Periodic paralysis Hypokalemic Thyrotoxic Hyperkalemic Other Central core disease Mitochondrial myopathy MELAS MERRF KSS PEO General Inflammatory myopathy Congenital myopathy

-
Lambert–eaton Myasthenic Syndrome
Wikipedia
This is attributed to the influx of calcium in response to these stimuli. [3] [2] On single-fiber examination, features may include increased jitter (seen in other diseases of neuromuscular transmission) and blocking. [3] Blood tests may be performed to exclude other causes of muscle disease (elevated creatine kinase may indicate a myositis , and abnormal thyroid function tests may indicate thyrotoxic myopathy ). Antibodies against voltage-gated calcium channels can be identified in 85% of people with EMG-confirmed LEMS. [3] Once LEMS is diagnosed, investigations such as a CT scan of the chest are usually performed to identify any possible underlying lung tumors. ... External links [ edit ] Classification D ICD - 10 : G73.1 , G70.8 ICD - 9-CM : 358.1 MeSH : D015624 DiseasesDB : 4030 External resources MedlinePlus : 000710 eMedicine : neuro/181 emerg/292 v t e Diseases of muscle , neuromuscular junction , and neuromuscular disease Neuromuscular- junction disease autoimmune Myasthenia gravis Lambert–Eaton myasthenic syndrome Neuromyotonia Myopathy Muscular dystrophy ( DAPC ) AD Limb-girdle muscular dystrophy 1 Oculopharyngeal Facioscapulohumeral Myotonic Distal (most) AR Calpainopathy Limb-girdle muscular dystrophy 2 Congenital Fukuyama Ullrich Walker–Warburg XR dystrophin Becker's Duchenne Emery–Dreifuss Other structural collagen disease Bethlem myopathy PTP disease X-linked MTM adaptor protein disease BIN1-linked centronuclear myopathy cytoskeleton disease Nemaline myopathy Zaspopathy Channelopathy Myotonia Myotonia congenita Thomsen disease Neuromyotonia / Isaacs syndrome Paramyotonia congenita Periodic paralysis Hypokalemic Thyrotoxic Hyperkalemic Other Central core disease Mitochondrial myopathy MELAS MERRF KSS PEO General Inflammatory myopathy Congenital myopathy v t e Paraneoplastic syndromes Endocrine Hypercalcaemia SIADH Zollinger–Ellison syndrome Cushing's syndrome Hematological Multicentric reticulohistiocytosis Nonbacterial thrombotic endocarditis Neurological Paraneoplastic cerebellar degeneration Encephalomyelitis Limbic encephalitis Opsoclonus Polymyositis Transverse myelitis Lambert–Eaton myasthenic syndrome Anti-NMDA receptor encephalitis Musculoskeletal Dermatomyositis Hypertrophic osteopathy Mucocutaneous reactive erythema Erythema gyratum repens Necrolytic migratory erythema papulosquamous Acanthosis nigricans Ichthyosis acquisita Acrokeratosis paraneoplastica of Bazex Extramammary Paget's disease Florid cutaneous papillomatosis Leser-Trélat sign Pityriasis rotunda Tripe palms Other Febrile neutrophilic dermatosis Pyoderma gangrenosum Paraneoplastic pemphigusPLEC, PREPL, CACNA1A, DAP, ACHE, GMPPB, SCLC1, DOK7, COL13A1, CHAT, HLA-DRB1, SYT1, RBM45, SYT2, WNK1, GORASP1, F11R, CEACAM4, CD274, EIF3K, CD2AP, CHRND, TNF, SOX2, HSPA5, SOX1, SLC18A3, RELA, DPAGT1, CEACAM6, ISG20, IL10, CXCL8, IL2RA, IL1B, ICAM1, PTEN

-
May–hegglin Anomaly
Wikipedia
External links [ edit ] Classification D ICD - 10 : D72.0 ICD - 9-CM : 288.2 OMIM : 155100 DiseasesDB : 29517 SNOMED CT : 250279001 External resources eMedicine : ped/1383 v t e Disorders of bleeding and clotting Coagulation · coagulopathy · Bleeding diathesis Clotting By cause Clotting factors Antithrombin III deficiency Protein C deficiency Activated protein C resistance Protein S deficiency Factor V Leiden Prothrombin G20210A Platelets Sticky platelet syndrome Thrombocytosis Essential thrombocythaemia DIC Purpura fulminans Antiphospholipid syndrome Clots Thrombophilia Thrombus Thrombosis Virchow's triad Trousseau sign of malignancy By site Deep vein thrombosis Bancroft's sign Homans sign Lisker's sign Louvel's sign Lowenberg's sign Peabody's sign Pratt's sign Rose's sign Pulmonary embolism Renal vein thrombosis Bleeding By cause Thrombocytopenia Thrombocytopenic purpura : ITP Evans syndrome TM TTP Upshaw–Schulman syndrome Heparin-induced thrombocytopenia May–Hegglin anomaly Platelet function adhesion Bernard–Soulier syndrome aggregation Glanzmann's thrombasthenia platelet storage pool deficiency Hermansky–Pudlak syndrome Gray platelet syndrome Clotting factor Haemophilia A/VIII B/IX C/XI von Willebrand disease Hypoprothrombinemia/II Factor VII deficiency Factor X deficiency Factor XII deficiency Factor XIII deficiency Dysfibrinogenemia Congenital afibrinogenemia Signs and symptoms Bleeding Bruise Haematoma Petechia Purpura Nonthrombocytopenic purpura By site head Epistaxis Haemoptysis Intracranial haemorrhage Hyphaema Subconjunctival haemorrhage torso Haemothorax Haemopericardium Pulmonary haematoma abdomen Gastrointestinal bleeding Haemobilia Haemoperitoneum Haematocele Haematosalpinx joint Haemarthrosis v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteins

-
Hereditary Pyropoikilocytosis
Wikipedia
External links [ edit ] Classification D OMIM : 266140 MeSH : C563004 C563004, C563004 v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteins v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline This article about a disease of the blood or immune system is a stub .SPTA1, SPTB, ALPL, ALPP, ASRGL1, ATHS, NAT10, PDLIM3, ATRNL1, CCL27, SLPI, SCN4A, KRAS, COL1A1, COL1A2, CACNA1S, EPB41, SOST, SYNE1, PANX1, PRDX5, CDKN2A, OTUD4, TUBGCP3, BRAF, PDXP, SOX17, APC, SCGB3A1, H19, CDX2, TLR2, BEST1, GATA4, SOX9, DMD, SFRP1, EGFR, PTHLH, MAPK3, PRRX1, PLP1, ENPP1, PRDX1, MLH1, HES1, IH

- Distal Myotilinopathy Orphanet
-
Progressive External Ophthalmoplegia With Mitochondrial Dna Deletions, Autosomal Recessive 3
OMIM
Muscle biopsy indicated a mitochondrial myopathy with up to 10% COX-negative ragged-red fibers. ... INHERITANCE - Autosomal recessive HEAD & NECK Face - Facial muscle weakness Eyes - External ophthalmoplegia, progressive - Ptosis CHEST Ribs Sternum Clavicles & Scapulae - Scapular winging ABDOMEN Gastrointestinal - Dysphagia MUSCLE, SOFT TISSUES - Muscle weakness, proximal - Muscle atrophy, mild, proximal - Mitochondrial myopathy - Myopathic features seen on EMG - Ragged red fibers seen on muscle biopsy - COX-negative fibers - Skeletal muscle shows mtDNA deletions - Decreased activities of mitochondrial-encoded respiratory chain complexes VOICE - Dysarthria LABORATORY ABNORMALITIES - Increased serum creatine kinase, mild - Increased serum lactate, mild MISCELLANEOUS - Onset in mid-forties - Two Finnish sisters have been reported (last curated August 2016) MOLECULAR BASIS - Caused by mutation in the nuclear-encoded mitochondrial thymidine kinase gene (TK2, 188250.0007 ) ▲ Close
- Juvenile Polymyositis Orphanet
-
Mevalonic Aciduria
Orphanet
A rare, very severe form of mevalonate kinase deficiency (MKD) characterized by dysmorphic features, failure to thrive, psychomotor delay, ocular involvement, hypotonia, progressive ataxia, myopathy, and recurrent inflammatory episodes.MVK, HMGCR, NLRP3, TNF, MEFV, IL1B, RAC1, TNFRSF1A, RHOA, SEC23B, NLRC4, ADA2, PMVK, SAA1, PSTPIP1, FDFT1, IL1A, IFNG, GRID2, MMAB

- Eosinophilic Fasciitis Orphanet
-
Phosphoglycerate Kinase 1 Deficiency
OMIM
Description Phosphoglycerate kinase-1 deficiency is an X-linked recessive condition with a highly variable clinical phenotype that includes hemolytic anemia, myopathy, and neurologic involvement. Patients can express 1, 2, or all 3 of these manifestations (Shirakawa et al., 2006). ... Shirakawa et al. (2006) reported a 33-year-old Japanese man with PGK1 deficiency manifesting as mental retardation and exertional myopathy, but without hemolytic anemia. ... In a patient with PGK1 deficiency manifest as myopathy (Sugie et al., 1989), Sugie et al. (1998) identified a mutation in the PGK1 gene (311800.0009). ... INHERITANCE - X-linked recessive HEAD & NECK Eyes - Retinal dystrophy (rare) - Loss of vision (rare) GENITOURINARY Kidneys - Renal failure may occur with myoglobinuria MUSCLE, SOFT TISSUES - Myopathy in approximately 45% of patients - Muscle cramps with exercise - Rhabdomyolysis - Exercise intolerance NEUROLOGIC Central Nervous System - Central nervous system involvement in approximately 50% of patients - Developmental delay - Mental retardation - Speech delay - Seizures - Hemiplegic migraines - Ataxia Behavioral Psychiatric Manifestations - Emotional instability HEMATOLOGY - Hemolytic anemia in approximately 60% of patients LABORATORY ABNORMALITIES - Myoglobinuria after exertion - Decreased hemoglobin - Increased serum bilirubin - Increased reticulocyte count - Decreased activity of phosphoglycerate kinase 1 MISCELLANEOUS - Highly variable phenotype - Variable age at onset (range infancy to adult) - Heterozygous females may exhibit variable degrees of enzyme deficiency MOLECULAR BASIS - Caused by mutation in the phosphoglycerate kinase 1 gene (PGK1, 311800.0002 ). ▲ Close
-
Lipodystrophy, Familial Partial, Type 6
OMIM
Description Familial partial lipodystrophy-6 (FPLD6) is characterized by abnormal subcutaneous fat distribution, with variable excess accumulation of fat in the face, neck, shoulders, axillae, back, abdomen, and pubic region, and reduction in subcutaneous fat of the lower extremities. Progressive adult-onset myopathy is seen in some patients, and there is variable association with diabetes, hypertriglyceridemia, low high-density lipoprotein (HDL) cholesterol, and hepatic steatosis (Zolotov et al., 2017). ... Zolotov et al. (2017) reported a 41-year-old woman and her 36-year-old brother, from a consanguineous Israeli Arab family, who exhibited marked symmetric accumulation of subcutaneous fat in the face, neck, axillae, and trunk, with loss of subcutaneous fat from the lower extremities and progressive symmetric myopathy during adulthood. In addition, the sister had diabetes and the brother had hepatic steatosis. ... In an Israeli Arab sister and brother with multiple symmetric lipomatosis, partial lipodystrophy, and myopathy, Zolotov et al. (2017) performed Sanger and exome sequencing and identified homozygosity for a nonsense mutation in the LIPE gene (E1035X; 151750.0003) that segregated with disease in the family. The authors concluded that LIPE deficiency in humans is associated with adult-onset progressive multiple symmetric lipomatosis, lower extremity lipodystrophy, and myopathy, along with metabolic abnormalities such as diabetes, hypertriglyceridemia, and hepatic steatosis.
-
Neuromuscular Junction Disease
Wikipedia
External links [ edit ] Classification D MeSH : D020511 External resources Orphanet : 98491 v t e Diseases of muscle , neuromuscular junction , and neuromuscular disease Neuromuscular- junction disease autoimmune Myasthenia gravis Lambert–Eaton myasthenic syndrome Neuromyotonia Myopathy Muscular dystrophy ( DAPC ) AD Limb-girdle muscular dystrophy 1 Oculopharyngeal Facioscapulohumeral Myotonic Distal (most) AR Calpainopathy Limb-girdle muscular dystrophy 2 Congenital Fukuyama Ullrich Walker–Warburg XR dystrophin Becker's Duchenne Emery–Dreifuss Other structural collagen disease Bethlem myopathy PTP disease X-linked MTM adaptor protein disease BIN1-linked centronuclear myopathy cytoskeleton disease Nemaline myopathy Zaspopathy Channelopathy Myotonia Myotonia congenita Thomsen disease Neuromyotonia / Isaacs syndrome Paramyotonia congenita Periodic paralysis Hypokalemic Thyrotoxic Hyperkalemic Other Central core disease Mitochondrial myopathy MELAS MERRF KSS PEO General Inflammatory myopathy Congenital myopathy







