-
Hydrocele Testis
Wikipedia
. ^ Ku u.a. 2001 KU, J. H.; KIM, M. E.; LEE, N. K.; PARK, Y. H.:The excisional, plication and internal drainage techniques: a comparison of the results for idiopathic hydrocele.In: BJU Int 87 (2001), Nr. 1, S. 82–4 ^ Fracchia, JA; Armenakas, NA; Kohan, AD (1998).FLT4, AKT1, PEX11B, GJC2, KLHL7, SOX18, PIGN, PIEZO1, ADAMTS3, VEGFC, RPL5, PTEN, PIK3CA, EZH2, MECOM, DIPK1A, CHIT1, TP63, VEGFA, EDN1, CYP4V2

-
Nevus Flammeus Nuchae
Wikipedia
. ^ MedlinePlus Encyclopedia : Stork bite External links [ edit ] Classification D OMIM : 163100 v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation Birthmark

-
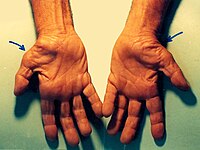
Carpal Tunnel Syndrome
OMIM
Further documentation of the improvement, which may obviate surgery, was presented by Ellis et al. (1982). They concluded that, since K(m) values of EGOT were identical in patients with and without CTS but with identical specific activities, CTS is a primary deficiency of B6, not a dependency state.TTR, AOC1, ADAMTS10, AEBP1, SMAD6, LTBP1, EFEMP1, HCP5, TTLL5, UMPS, ADAMTS17, B2M, PMP22, CCN2, IL1B, PSMD9, CYP19A1, GSTT1, COL5A1, PGR, SLX1A-SULT1A3, SEMG1, PDGFRB, SULT1A4, MIR106A, MAPK3, PDGFRA, SERPINE1, PTGS2, ROS1, CCL2, DHDDS, SLC6A2, SULT1A3, CHPT1, TGFB1, TGFBR1, GSTK1, NT5E, VDR, VEGFA, HDAC4, ADAMTS5, SLCO6A1, INTU, HPGDS, SEMA5B, OLR1, MMP3, NDUFB2, CECR, ELK3, ACE, CSH2, CSH1, COMT, COL11A1, ERCC8, CASP8, COX2, CALCR, BGN, AMH, ABCD1, ALB, AKT1, ACAN, EPHB1, ESR1, FAP, GHR, MPZ, MMP10, MMP9, MMP1, MIP, IL6R, IL1RN, IGF1, TNC, HSPA9, HLA-DRB3, HLA-DPB1, HLA-DPA1, GSTP1, GSTM1, MTCO2P12
-
Hyperinsulinemic Hypoglycemia, Familial, 7
OMIM
Quintens et al. (2008) noted that repression of certain ubiquitously expressed housekeeping proteins is necessary in pancreatic beta cells, in order to prevent the insulin toxicity that might result from exocytosis under conditions when circulating insulin is unwanted, citing low-K(m) hexokinases (see HK1, 142600) and monocarboxylic acid transporters (MCTs) as examples.
- Microvillous Inclusion Disease Wikipedia
- Pmm2 Deficiency Wikipedia
-
Anomic Aphasia
Wikipedia
.; Cooper-Pye, E.; Hodges, JR.; Patterson, K. (Aug 2008). "Anomia: a doubly typical signature of semantic dementia". ... PMID 3978406 . ^ Miller, N; Willmes, K; De Bleser, R (2000). "The psychometric properties of the English language version of the Aachen Aphasia Test (EAAT)". ... PMID 21636820 . ^ Martin, N; Fink, R; Renvall, K; Laine, M (Nov 12, 2006). "Effectiveness of contextual repetition priming".
-
Paraphilic Infantilism
Wikipedia
Retrieved 2006-03-07 . ^ a b Kise, K.; Nguyen, M. (2011). "Adult Baby Syndrome and Gender Identity Disorder". ... ISBN 978-0-313-32968-5 . ^ a b c d Stekel 1952 , pp. 143–144. ^ a b Hawkinson, K., & Zamboni, B. D. (2014). "Adult Baby/Diaper Lovers: An Exploratory Study of an Online Community Sample". ... Continuum International Publishing Group . p. 255. ISBN 978-0-8264-1026-9 . ^ Freund K; Blanchard R (1993). "Erotic target location errors in male gender dysphorics, paedophiles, and fetishists".
-
Antenatal Depression
Wikipedia
Retrieved 2019-10-24 . ^ a b c Kroenke K, Spitzer RL, Williams JB (September 2001). ... S2CID 24565503 . ^ Misri S, Kendrick K, Oberlander TF, Norris S, Tomfohr L, Zhang H, Grunau RE (April 2010). ... PMID 20416145 . ^ Gress-Smith JL, Luecken LJ, Lemery-Chalfant K, Howe R (May 2012). "Postpartum depression prevalence and impact on infant health, weight, and sleep in low-income and ethnic minority women and infants".
-
Encephalitis Lethargica
Wikipedia
At the time, doctors attributed her encephalitis to having contracted influenza during the 1918 pandemic. [38] References [ edit ] ^ Economo's disease at Who Named It? ^ von Economo, K. (May 10, 1917). "Die Encephalitis lethargica". ... S2CID 45138249 . ^ a b McCall, Sherman; Vilensky, Joel A; Gilman, Sid; Taubenberger, Jeffery K (May 2008). "The relationship between encephalitis lethargica and influenza: A critical analysis" . ... PMID 18569452 . ^ a b Haeman, Jang; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, Richard J. (2009).

-
Streptococcal Pharyngitis
Wikipedia
It is, however, the leading cause of acquired heart disease in India, sub-Saharan Africa, and some parts of Australia. [8] Complications arising from streptococcal throat infections include: Acute rheumatic fever [11] Scarlet fever [36] Streptococcal toxic shock syndrome [36] [37] Glomerulonephritis [38] PANDAS syndrome [39] [40] [41] Peritonsillar abscess [8] Cervical lymphadenitis [8] Mastoiditis [8] The economic cost of the disease in the United States in children is approximately $350 million annually. [8] Epidemiology Pharyngitis , the broader category into which Streptococcal pharyngitis falls, is diagnosed in 11 million people annually in the United States. [10] It is the cause of 15–40% of sore throats among children [7] [10] and 5–15% in adults. [8] Cases usually occur in late winter and early spring. [10] References ^ a b c d e f g h i j k l m n o p "Is It Strep Throat?" . ... S2CID 8625679 . ^ a b c d e f g h i j k l m n o p q r Shulman, ST; Bisno, AL; Clegg, HW; Gerber, MA; Kaplan, EL; Lee, G; Martin, JM; Van Beneden, C (Sep 9, 2012). ... PMID 26785402 . ^ a b c d e f g h i j k l m n o p q r s t u v w x Choby BA (March 2009).

-
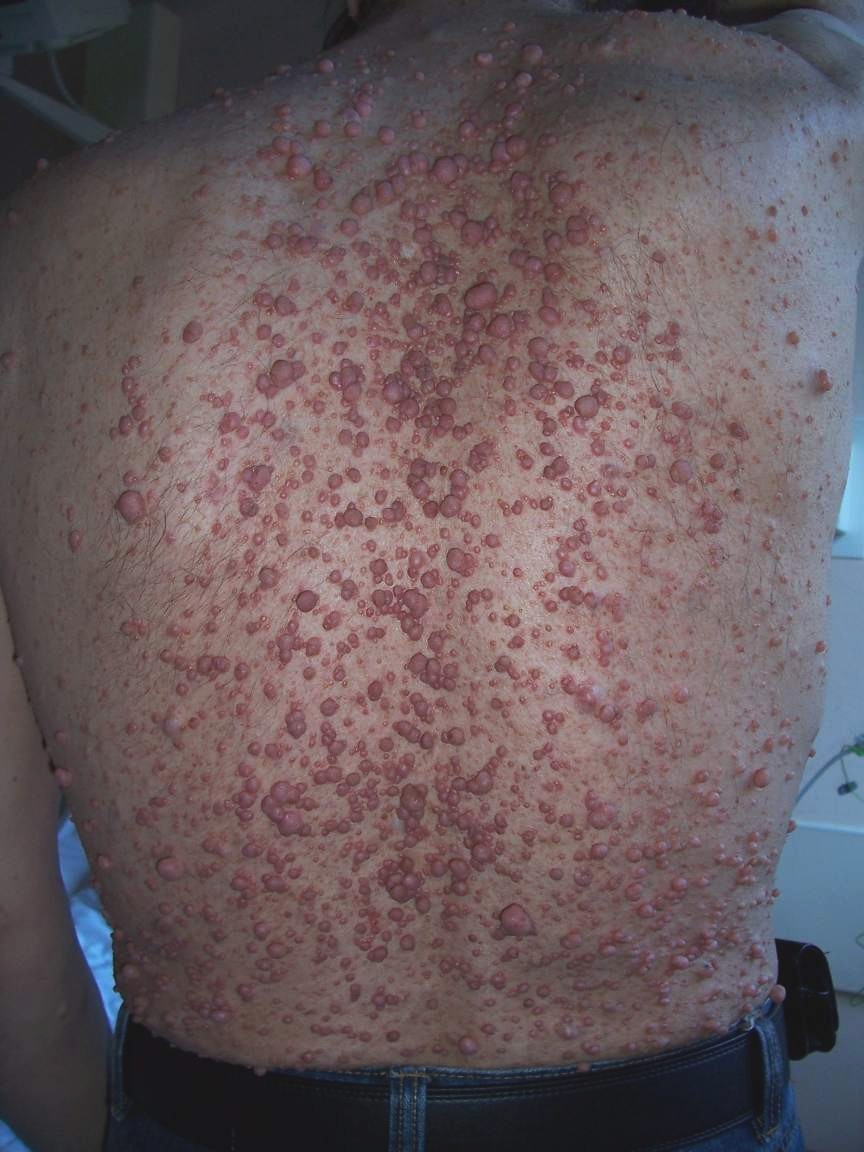
Neurofibroma
Wikipedia
. ^ a b "Case Based Pediatrics For Medical Students and Residents: Chapter XVIII.11. Neurofibromatosis", by Vince K. Yamashiroya, MD. August, 2002. Department of Pediatrics, University of Hawaii John A. ... Archives of Orthopaedic and Trauma Surgery . 127 (8): 709–12. doi : 10.1007/s00402-007-0303-1 . PMID 17377797 . ^ Iino K, Matsumoto Y, Endo M, et al. (2006). ... PMID 21319287 . ^ Yamamoto M, Yamauchi T, Okano K, Takahashi M, Watabe S, Yamamoto Y (March 2009).NF1, SUZ12, KIT, NF2, SPRED1, CDKN2A, PDGFB, TP53, SUFU, SMARCB1, SDHC, SDHB, EGFR, S100B, PDGFRA, S100A1, MDK, NRG1, EGF, SMUG1, VEGFA, FGF2, MDM2, SOX9, PDGFRB, CCND1, FN1, TOP2A, BRAF, CDKN2B-AS1, STAT3, CTNNB1, SST, EED, HMGA2, SYK, CNTN2, FGF23, VDR, TGFA, AURKA, NKX2-1, CXCR4, WT1, TLE1, TGFB1, ALDH1A1, RGS6, SPRY1, H3P23, TMED7-TICAM2, CCR2, MIR339, MIR34A, MIR210, MIR21, MIR10B, TICAM2, GADL1, THSD7A, AZIN2, ARID1B, ADAP2, TMED7, CD274, PDCD4, BRD4, KANK1, SNF8, DCTN6, ZNRD2, CIB1, NES, PTN, SOAT1, DCX, GHSR, GEM, FOLH1, MLANA, EZH2, EVI2B, ERBB2, EPHB2, EFNA3, DUSP5, CSF1, GSTP1, COL3A1, CNP, CDKN2B, CDKN1B, CDK4, CD44, CD38, RUNX1, ATRX, ATM, CXCR3, HGF, SMARCA2, PPARD, CXCL12, CCL2, RASA1, PTPN11, AR, PTEN, PSMD9, MAPK3, MAPK1, PRKAR1A, PMP22, IFI27, PLG, PGR, PDGFA, SERPINE1, NGF, MVD, MMP13, LEP, CXCL10, IFNA2, H3P10
-
Periventricular Leukomalacia
Wikipedia
Robbin's Review of Pathology. 2nd ed. pp. 116, 121 ^ Tsukimori, K; Komatsu, H; Yoshimura, T; Hikino, S; Hara, T; Wake, N; Nakano, H (August 2007). ... ISSN 0022-3565 . PMID 10027860 . ^ Yokochi, K (March 2001). "Gait patterns in children with spastic diplegia and periventricular leukomalacia". ... Accessed November 27, 2008 External links [ edit ] Classification D ICD - 10 : P91.2 ICD - 9-CM : 779.7 MeSH : D007969 DiseasesDB : 9868 External resources MedlinePlus : 007232 eMedicine : ped/1773 v t e Conditions originating in the perinatal period / fetal disease Maternal factors complicating pregnancy, labour or delivery placenta Placenta praevia Placental insufficiency Twin-to-twin transfusion syndrome chorion / amnion Chorioamnionitis umbilical cord Umbilical cord prolapse Nuchal cord Single umbilical artery presentation Breech birth Asynclitism Shoulder presentation Growth Small for gestational age / Large for gestational age Preterm birth / Postterm pregnancy Intrauterine growth restriction Birth trauma scalp Cephalohematoma Chignon Caput succedaneum Subgaleal hemorrhage Brachial plexus injury Erb's palsy Klumpke paralysis Affected systems Respiratory Intrauterine hypoxia Infant respiratory distress syndrome Transient tachypnea of the newborn Meconium aspiration syndrome Pleural disease Pneumothorax Pneumomediastinum Wilson–Mikity syndrome Bronchopulmonary dysplasia Cardiovascular Pneumopericardium Persistent fetal circulation Bleeding and hematologic disease Vitamin K deficiency bleeding HDN ABO Anti-Kell Rh c Rh D Rh E Hydrops fetalis Hyperbilirubinemia Kernicterus Neonatal jaundice Velamentous cord insertion Intraventricular hemorrhage Germinal matrix hemorrhage Anemia of prematurity Gastrointestinal Ileus Necrotizing enterocolitis Meconium peritonitis Integument and thermoregulation Erythema toxicum Sclerema neonatorum Nervous system Perinatal asphyxia Periventricular leukomalacia Musculoskeletal Gray baby syndrome muscle tone Congenital hypertonia Congenital hypotonia Infections Vertically transmitted infection Neonatal infection rubella herpes simplex mycoplasma hominis ureaplasma urealyticum Omphalitis Neonatal sepsis Group B streptococcal infection Neonatal conjunctivitis Other Miscarriage Perinatal mortality Stillbirth Infant mortality Neonatal withdrawalTNF, MBP, AARS2, TPI1, ARID1A, PLA2G6, SON, DDHD2, RPS6KC1, PMM2, PC, NOTCH1, TMCO1, DLL4, MCOLN1, RNF113A, MPLKIP, ARID2, RBPJ, CLCN4, DOCK6, TBCK, GTF2E2, GTF2H5, ERCC3, EOGT, ERCC2, ARHGAP31, IL6, STIP1, NES, SQSTM1, NOP53, ZNF260, KRT20, ARHGEF5, ANC, SERPINH1, TFPI, VEGFA, IL10, COL4A1, CRP, ACE, ECHS1, EPO, HOXD13, IFIT3, IL2, CXCL8, IL18, TNFRSF1A, MYT1, PEX1, SFTPD, SLC1A2, SOD2, SOD3, SOX2, MS4A1, TLR2, H3P7

-
Uterine Atony
Wikipedia
Most women with mild to moderate anemia, however, resolve the anemia sufficiently rapidly with oral iron alone and do not need parenteral iron. [1] [2] Prognosis [ edit ] Women with a history of PPH have a 2 to 3 times higher risk of PPH in their following pregnancies. [36] [1] [37] References [ edit ] ^ a b c d e f g h i j k l m n o p q r s t u v Gill P, Patel A, Van Hook JW (2020). ... PMC 1298111 . PMID 11089490 . ^ a b Belghiti K, Kayem G, Dupont C, Rudigoz RC, Bouvier-Colle MH, Deneux-Tharaux C (2011-12-21). ... S2CID 2190885 . ^ Oberg AS, Hernandez-Diaz S, Palmsten K, Almqvist C, Bateman BT (2014). "Patterns of recurrence of postpartum hemorrhage in a large population-based cohort" .
-
Spectrum Disorder
Wikipedia
CS1 maint: multiple names: authors list ( link ) ^ Tienari P, Wynne LC, Läksy K, et al. (September 2003). "Genetic boundaries of the schizophrenia spectrum: evidence from the Finnish Adoptive Family Study of Schizophrenia". ... PMID 16960654 . ^ Angst J, Merikangas K (August 1997). "The depressive spectrum: diagnostic classification and course". ... PMID 16330723 . ^ Lara DR, Pinto O, Akiskal K, Akiskal HS (August 2006). "Toward an integrative model of the spectrum of mood, behavioral and personality disorders based on fear and anger traits: I.
-
Ketosis
Wikipedia
Ketosis Other names Ketonemia Ketone bodies : acetone, acetoacetic acid, and beta-hydroxybutyric acid Pronunciation / k ɪ ˈ t oʊ s ɪ s / Specialty Endocrinology Ketosis is a metabolic state characterized by elevated levels of ketone bodies in the blood or urine. [1] Physiologic ketosis is a normal response to low glucose availability, such as low-carbohydrate diets or fasting , that provides an additional energy source for the brain in the form of ketones. ... Retrieved 30 September 2019 . ^ a b c d Mattson MP, Moehl K, Ghena N, Schmaedick M, Cheng A (2018). ... PMID 14769489 . ^ Fukao T, Mitchell G, Sass JO, Hori T, Orii K, Aoyama Y (2014). "Ketone body metabolism and its defects".ACAT1, PAX4, INS, SLC16A1, GCK, ABCC8, ITPR3, IL6, GYS2, POLG2, GK, GHSR, POLG, HNF1A, MLYCD, FBP1, PTPN22, EIF2AK3, DBT, PDX1, RRM2B, ACSF3, SLC5A2, TWNK, PHKG2, MMAA, ATP5F1D, BCKDHA, BCKDHB, ACADM, PHKA2, CA5A, IVD, MMAB, MCCC2, KCNJ11, SLC25A4, FGF21, GLP1R, SLC30A8, SLC30A10, GCG, LEP, MCTS1, MCAT, CMA1, INSR, SREBF1, SOAT1, SLC2A2, PTEN, MAPK3, PPARD, ACAA1, TNF, CHPT1, ZGLP1, ZFP57, TAS2R12P, NRSN1, RBM45, SOCS4, EHMT1, APOBR, UBL4A, ATG14, CHP1, TPPP, TMED2, ACAA2, RGS6, CD163, POMC, MARK1, PIK3CG, PTK2B, DMD, CYP7A1, CYP2E1, CYC1, CPT2, CPT1A, CORT, CDR2, CD9, CCK, BMP4, BGLAP, APOE, ALDH2, AKT2, AKT1, AHSG, ADH1B, ACP1, DPP4, G6PD, PIK3CD, GAD1, PIK3CB, PIK3CA, PAX6, OXCT1, ORM1, TNFRSF11B, NHS, CD46, LPL, IL1B, IFNG, IDE, HNF4A, HLCS, HLA-DRB1, HIF1A, HADHB, GCLC, GAD2, H3P19

-
Group A Streptococcal Infection
Wikipedia
CS1 maint: archived copy as title ( link ) ^ Howard, SJ; Stoker, K; Foster, K (16 June 2015). "Public health management of group A streptococcal infection in mother-baby pairs in England; a case series review" . ... E.; Roberson, A.; Cieslak, P. R.; Lynfield, R.; Gershman, K.; Craig, A.; Albanese, B. A.; Farley, M.

-
Drug Interaction
Wikipedia
The former acts on cardiac fibres and its effect is increased if there are low levels of potassium (K) in blood plasma. Furosemide is a diuretic that lowers arterial tension but favours the loss of K + . ... PMID 7568331 . ^ Palmer, Adam C.; Sorger, Peter K. (2017-12-14). "Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy" . ... PMID 19109115 . ^ Haider SI, Johnell K, Thorslund M, Fastbom J (December 2007). ... PMID 18184532 . ^ Haider SI, Johnell K, Weitoft GR, Thorslund M, Fastbom J (January 2009).
-
Rhinitis
Wikipedia
For allergic rhinitis, intranasal corticosteroids are recommended. [45] For severe symptoms intranasal antihistamines may be added. [45] Pronunciation and etymology [ edit ] Rhinitis is pronounced / r aɪ ˈ n aɪ t ɪ s / , [46] while coryza is pronounced / k ə ˈ r aɪ z ə / . [47] Rhinitis comes from the Ancient Greek ῥίς rhis , gen .: ῥινός rhinos "nose". ... PMID 27227021 . ^ Eriksson J, Bjerg A, Lötvall J, Wennergren G, Rönmark E, Torén K, Lundbäck B (November 2011). "Rhinitis phenotypes correlate with different symptom presentation and risk factor patterns of asthma". ... PMID 21764573 . ^ Scherer Hofmeier K, Bircher A, Tamm M, Miedinger D (April 2012).

-
Lymphatic Filariasis
Wikipedia
Archived from the original on 2016-10-12. ^ a b c d e f g h i j k l m n o p q r s t u "Lymphatic filariasis Fact sheet N°102" . ... Univ Puerto Rico Med J . 22 : 187–193. [ verification needed ] ^ Saladin K (2007). Anatomy & Physiology: The Unity of Form and Function . ... Archived from the original on 2017-07-13. ^ Deribe K, Tomczyk S, Tekola-Ayele F (2013).GPT2, THAS, IL10, HSPD1, TLR9, TLR2, LMLN, GGTLC1, TMPRSS13, NARS2, RBM33, TLR1, HSPA14, NBEAL2, HERPUD1, NR0B2, VEGFA, TXN, TNFRSF1B, TLR4, CISH, CTLA4, MSMB, EDN1, FOXC2, FLT4, HGF, IGHG3, IL3, MST1, TGFB1, NARS1, PRL, PTEN, PTPN6, SLA, SMS, LOC102724197














