This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( August 2015 ) Post-vagotomy diarrhea Course of vagus nerve Specialty Gastroenterology Post-vagotomy diarrhea is a form of diarrhea which occurs in 10% of people after a truncal vagotomy , which can range from severe to debilitating in approximately 2% to 4% of patients. [1] However, the occurrence of post-vagotomy diarrhea is significantly reduced after proximal selective vagotomy, specifically when celiac and hepatic branches of the vagus are retained. [1] Contents 1 Diagnosis 2 Treatment 3 References Diagnosis [ edit ] This section is empty.
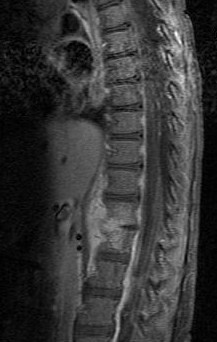
Spondylodiscitis Specialty Rheumatology Spondylodiscitis is a combination of discitis ( inflammation of one or more intervertebral disc spaces ) and spondylitis (inflammation of one or more vertebrae ), the latter generally involving the areas adjacent to the intervertebral disc space. [1] Contents 1 Causes 2 Diagnosis 3 References 4 External links Causes [ edit ] Spondylodiscitis is the most common complication of sepsis or local infection, usually in the form of an abscess. [2] The main causative organisms are staphylococci and Mycobacterium tuberculosis , but potential organisms include a large number of bacteria, fungi, zoonoses. [2] Spondylodiscitis frequently develops in immunocompromised individuals, such as by a cancer, infection, or by immunosuppressive drugs used for organ transplantations. [2] Diagnosis [ edit ] The main methods to diagnose a spondylodiscitis are magnetic resonance imaging (MRI), biopsy and microbiological tests such as PCR to determine an infectious cause. [2] References [ edit ] ^ Page 147 in: Hinchcliffe, Ronald; Fritz Hefti; Jundt, Gernot; Freuler, F. (2007).
In children whether to bed rest or move a little is decided on an individual basis, depending on the site and severity of the discitis. References [ edit ] ^ Page 147 in: Hinchcliffe, Ronald; Fritz Hefti; Jundt, Gernot; Freuler, F. (2007).
Fitzpatrick's Dermatology in General Medicine . (6th ed.). Page 994. McGraw-Hill. ISBN 0-07-138076-0 . ^ James, William D.; Berger, Timothy G.; et al. (2006).
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( September 2017 ) Benign metastasizing leiomyoma Specialty Oncology Benign metastasizing leiomyoma is a rare condition characterized by the growth of uterine leiomyoma in the other regions especially the lungs . [1] [2] [3] [4] References [ edit ] ^ Fu, Yili; Li, Hui; Tian, Bo; Hu, Bin (2012).
Please help improve it or discuss these issues on the talk page . ( Learn how and when to remove these template messages ) The topic of this article may not meet Wikipedia's general notability guideline .
Clinical Features Chilblain lupus, a rare cutaneous form of systemic lupus erythematosus (152700), was first described by Jonathan Hutchinson (1888). ... The family study suggested a highly penetrant trait with autosomal dominant inheritance. Chilblain lupus occurs predominantly in adult women and has only rarely been described in children. ... History For a review of the career and clinical observations of Hutchinson, who first described chilblain lupus, see McKusick (1952, 2005). INHERITANCE - Autosomal dominant CARDIOVASCULAR Vascular - No Raynaud phenomenon SKELETAL - Arthralgias (knees and shoulders) SKIN, NAILS, & HAIR Skin - Painful bluish-red papules or nodules (fingers, toes, nose, cheek, ears) - Cutaneous ulcers - Healed areas are atrophic and hypopigmented - No cutaneous photosensitivity Skin Histology - Deep inflammatory perivascular infiltrate with granular deposits of immunoglobulins and complement along basement membrane Nails - Subungual lesions (in some patients) IMMUNOLOGY - Antinuclear antibody present (in some patients) MISCELLANEOUS - Environmental triggers - cold and wet exposure - Onset in early childhood - Allelic to Aicardi-Goutieres syndrome ( 225750 ) MOLECULAR BASIS - Caused by mutation in the 3-prime repair exonuclease-1 gene (TREX1, 606609.0005 ) ▲ Close
Lupus pernio Cutaneous lesions of sarcoidosis (lupus pernio). Red-to-purple indurated plaques and nodules affecting the nose and cheeks. Specialty Dermatology Lupus pernio is a chronic raised indurated (hardened) lesion of the skin, often purplish in color. It is seen on the nose , ears , cheeks , lips , and forehead . It is pathognomonic of sarcoidosis . [1] : 701 The name "lupus pernio" is a misnomer , as microscopically this disease shows granulomatous infiltration and does not have features of either lupus or pernio . [2] Lupus pernio is associated with poor outcomes and lower rates of resolution. Lupus pernio and erythema nodosum are cutaneous manifestation of sarcoidosis, may suggest this disease as a cause of an associated dilated cardiomyopathy, especially with heart block, intraventricular conduction delay, or ventricular tachycardia. See also [ edit ] Sarcoidosis List of cutaneous conditions References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2011).
A rare monogenic form of cutaneous lupus erythematosus characterized by infantile or childhood onset of cold-induced erythematous papules or plaques predominantly on the fingers, toes, nose, cheeks, and ears. Recurrent ulceration of the lesions may lead to necrotic tissue destruction and mutilation. Patients may experience ischemia of the affected acral regions. Histological findings include cutaneous perivascular inflammatory infiltrates with deposits of immunoglobulins or complement.
A rare, chronic cutaneous lupus erythematosus disease characterized by red or violaceous, initially pruritic (evolving to painful) papules and plaques located on acral areas (especially dorsal aspects of fingers and toes, while the nose and ear involvement is uncommon), exacerbated by cold and damp conditions, with fissuring and ulceration occasionally observed. Coexistence of discoid lupus erythematosus lesions elsewhere on the body and occasional progression to systemic lupus erythematosus may be associated. Histological examination and direct immunofluorescence studies reveal nonspecific inflammatory lupus erythematosus changes while results of cryoglobulin and cold agglutinin studies are negative.
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( September 2012 ) Qazi–Markouizos syndrome Other names Dysharmonic skeletal maturation-muscular fiber disproportion syndrome [1] Qazi–Markouizos syndrome is a rare hereditary condition characterized by non-progressive, congenital hypotonia , severe intellectual disability, an increased proportion of type 2 muscle fibers , which additionally exhibited increased size, as well as dysharmonic skeletal maturation . [2] [3] To date, the molecular mechanism of Qazi–Markouizos syndrome, which is also known as Puerto Rican infant hypotonia syndrome, [4] remains unknown.
Clinical Features Qazi et al. (1994) reported a seemingly distinct and 'new' syndrome in 3 unrelated Puerto Rican boys. Features were marked, central, nonprogressive hypotonia, chronic constipation, severe psychomotor retardation, seizures or abnormal electroencephalogram or both, abnormal dermatoglyphics, delayed bone age, dysharmonic skeletal maturation, and preponderance and larger size of type 2 muscle fibers. Additional findings included narrow and high-arched palate, prominent nasal root, long philtrum, distended abdomen, and drooling from open mouth. Two of the boys had undescended testes, hypertelorism, and tapered fingers. Birth weight, postnatal physical growth, and head size were average. Photographs of the 3 demonstrated the open mouth and other facial features.
A rare, genetic, syndromic intellectual disability disorder characterized by non-progressive, congenital, marked, central hypotonia, severe psychomotor delay and intellectual disability, chronic constipation, distended abdomen, abnormal dermatoglyphics, delayed and dysharmonic skeletal maturation, and preponderance of type 2 larger-sized muscle fibers. Additional features include narrow and high-arched palate, prominent nasal root, long philtrum, and open mouth with drooling, as well as variably present cryptorchidism, hypertelorism, and tapered fingers. Seizures and/or an abnormal electroencephalograph may also be assoicated. There have been no further descriptions in the literature since 1994.
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( April 2014 ) Binocular Dysphoria is a hypothesized condition where the brain adapts to an alternative way of perceiving depth cues. 3-D films , televisions, virtual reality headsets, and other devices simulate the experience of three dimensions through stereoscopic techniques, presenting slightly different images to the left and right eyes.
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( April 2016 ) Deep dermatophytosis Other names Disseminated granulomatous dermatophytosis Deep dermatophytosis is a rare condition in which dermatophytes invades the deep dermis , subcutis or even internal organs .
A rare mycosis characterized by severe, potentially life-threatening dermal and subcutaneous tissue invasion by dermatophytes. Dissemination to lymph nodes is frequent, but the infection may also occasionally spread to the central nervous system. Cutaneous signs and symptoms include erythema, desquamation, itching, nodules, plaques, or ulceration. The majority of deep dermatophytoses develop in immunocompromised patients.
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( November 2019 ) This article includes a list of references , related reading or external links , but its sources remain unclear because it lacks inline citations .
Schmitt et al. (1982) described a family in which 5 females and 3 males over 3 generations had bilateral, symmetric, nonopposable triphalangeal thumbs and radial hypoplasia. Affected males had first-degree hypospadias and all affected persons had anterior maxillary diastema. No male-to-male transmission was observed; however, transmission to only 2 of 4 daughters by an affected male favors autosomal (as opposed to X-linked) dominant inheritance. Limbs - Nonopposable triphalangeal thumbs - Radial hypoplasia Inheritance - Autosomal dominant GU - First-degree hypospadias in males Facies - Anterior maxillary diastema ▲ Close
Schmitt Gillenwater Kelly syndrome Other names Radial hypoplasia-triphalangeal thumbs-hypospadias-maxillary diastema syndrome Schmitt Gillenwater Kelly syndrome has an autosomal dominant pattern of inheritance . Schmitt Gillenwater Kelly syndrome is a rare autosomal dominant [1] congenital disorder consisting of radial hypoplasia , triphalangeal thumbs , hypospadias , and maxillary diastema . [1] [2] References [ edit ] ^ a b Schmitt E, Gillenwater JY, Kelly TE (1982). "An autosomal dominant syndrome of radial hypoplasia, triphalangeal thumbs, hypospadias, and maxillary diastema".
Sequence analysis of CHST3 is performed first. If no pathogenic variants or only a single pathogenic variant have been identified and the clinical-radiographic index of suspicion is high for a CHST3 -related skeletal dysplasia, consider doing gene-targeted deletion/duplication analysis. ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... Nomenclature In 1950, Dr LJ Larsen described autosomal dominant Larsen syndrome, now known to be caused by pathogenic variants in FLNB , the gene encoding filamin B [Bicknell et al 2007]. ... Following the delineation of autosomal dominant Larsen syndrome, several reports of "autosomal recessive Larsen syndrome" and other similar disorders were published. ... Because the brothers were reported to be half-sibs, autosomal dominant inheritance was suspected and no link was made to autosomal recessive Larsen syndrome.
CHST3 -related skeletal dysplasia is a genetic condition characterized by bone and joint abnormalities that worsen over time. Affected individuals have short stature throughout life, with an adult height under 4 and a half feet. Joint dislocations, most often affecting the knees, hips, and elbows, are present at birth (congenital). Other bone and joint abnormalities can include an inward- and upward-turning foot (clubfoot ), a limited range of motion in large joints, and abnormal curvature of the spine . The features of CHST3 -related skeletal dysplasia are usually limited to the bones and joints; however, minor heart defects have been reported in a few affected individuals.
LMS is inherited in an autosomal dominant manner. Although most probands have the disorder as a result of a de novo NOTCH3 pathogenic variant, affected parent-child pairs have been reported. ... Sequence analysis of NOTCH3 is performed first, followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found. ... Marfan syndrome is inherited in an autosomal dominant manner and is caused by mutation of FBN1 . ... Noonan syndrome is inherited in an autosomal dominant manner. Genes known to be associated with the disorder include PTPN11 , SOS1 , RAF1 , RIT1 , and KRAS . ... NF1 is inherited in an autosomal dominant manner and is caused by mutation of NF1 .
Learn more about the gene associated with Lateral meningocele syndrome NOTCH3 Inheritance Pattern This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
A rare genetic neurological disorder characterized by multiple lateral meningoceles, distinctive facial dysmorphism (including hypertelorism, downslanting palpebral fissures, posteriorly rotated ears, micrognathia, and high, narrow palate, among others), and skeletal abnormalities (e. g. vertebral anomalies, wormian bones, short stature, and scoliosis). Multiple additional features may present, such as conductive hearing impairment, hypotonia, and connective tissue and urogenital abnormalities. Cognition is usually normal.
Lateral meningocele syndrome Other names Lehman syndrome [1] Lateral meningocele syndrome is inherited in an autosomal dominant manner The lateral meningocele syndrome is a very rare skeletal disorder with facial anomalies, hypotonia and meningocele-related neurologic dysfunction. [2] Contents 1 Presentation 2 Genetics 3 Diagnosis 4 Treatment 5 History 6 References 7 External links Presentation [ edit ] Facial features found in this syndrome include dolichocephaly hypertelorism ptosis microretrognathia high arched palate long flat philtrum low set ears Non facial features of this syndrome include hyperextensibility hypotonia lateral meningoceles The lateral meningoceles are a common finding in this syndrome. ... Genetics [ edit ] This syndrome appears to be inherited in an autosomal dominant fashion. Molecular analyses suggest that the causative mutations cause a truncation of the protein. ... You can help by adding to it . ( September 2017 ) History [ edit ] This syndrome was first described by Lehman et al. in 1977. [4] This paper described a 14-year-old girl with a number of unusual findings.
When SRY is translocated to another chromosome or when fertility is preserved, sex-limited autosomal dominant inheritance is observed. Autosomal dominant inheritance has been documented for familial cases thought to be caused by CNV in or around SOX9 . ... Small duplication or triplication of the promoter region of SOX9 , a balanced chromosomal translocation involving the 17q24.3 region, or duplication of SOX9 (in mosaic or nonmosaic form) [Huang et al 1999, Refai et al 2010, Cox et al 2011, Vetro et al 2011, Xiao et al 2013, Lee et al 2014, Kim et al 2015, Vetro et al 2015] SOX3 . Microdeletions just upstream of the open reading frame of SOX3 [Sutton et al 2011] or microduplications in SOX3 [Sutton et al 2011, Moalem et al 2012] Molecular Genetic Testing Strategy In an individual with ambiguous genitalia in whom no chromosome study has been performed One testing option is to perform karyotype with FISH for SRY first. If the karyotype is normal 46,XX and FISH for SRY is negative, proceed to chromosomal microarray (CMA). An alternative option is to perform CMA first, which will give information about the sex chromosome complement, the presence of SRY , and other copy number variants of clinical relevance. ... One report of a small duplication in the SOX9 promoter region in a newborn with a 46,XX karyotype and ambiguous genitalia states that the duplication was found not only in the proband's 46,XY fertile father and phenotypically normal male brothers but also in his paternal 46,XX (fertile) grandmother [Benko et al 2011].
In some cases, gonadal surgery can be performed to remove partial or whole female genitalia. This may be followed by plastic and reconstructive surgery to make the individual appear more externally male. [25] Conversely, the individual may wish to become more feminine and feminizing genitoplasty can be performed to make the ambiguous genitalia appear more female. [26] Hormonal therapy may also aid in making an individual appear more male or female. [25] [26] Testosterone [ edit ] Testosterone At puberty, most affected individuals require treatment with the male sex hormone testosterone to induce development of male secondary sex characteristics such as facial hair and deepening of the voice (masculinization). ... Retrieved 2020-09-06 . ^ Andersson, M.; Page, D. C.; de la Chapelle, A. (1986-08-15).
A rare disorder of sex development (DSD) associated with a 46, XX karyotype and characterized by male external genitalia, ranging from normal to atypical with associated testosterone deficiency. Epidemiology The estimated prevalence is 1/20,000 males. Clinical description The clinical phenotype is variable, with features that include: normal male external to atypical genitalia, undescended testes with absent Müllerian structures and infertility. Presentation depends on the presence of the SRY gene (sex determining region of the Y chromosome). SRY positive cases (80-90%) are usually otherwise normal men who present after puberty with short stature, normal pubic hair and penile size but small testes, gynecomastia and azoospermia-related sterility. Undescended testes and hypospadias are also reported. There are usually no concerns about gender role and identity.
Sequence analysis of the gene of interest is performed first, followed by gene-targeted deletion/duplication analysis if only one or no pathogenic variant is found. In individuals with harlequin ichthyosis, analysis of ABCA12 should be performed first. In individuals with ARCI and without harlequin presentation at birth, analysis of TGM1 should be performed first. ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... Nomenclature Historically, the term "lamellar ichthyosis" was used to describe any individual with ARCI, and even rare cases of autosomal dominant ichthyosis, regardless of whether erythroderma was present. ... Individuals with autosomal dominant epidermolytic ichthyosis virtually never present with a collodion membrane at birth.
Both occur particularly with liver metastasis , and either symptom may be the first manifestation of the disease. The flushing that occurs in medullary thyroid carcinoma is indistinguishable from that associated with carcinoid syndrome. ... Hereditary medullary thyroid cancer is inherited as an autosomal dominant trait, meaning that each child of an affected parent has a 50% probability of inheriting the mutant RET proto-oncogene from the affected parent. ... For those in the highest risk group, surgery is recommended in the first year of life. In lower risk cases surgery may be delayed up to the age of ten years, the precise timing depending on the mutation and other factors. ... Vandetanib , trade name Caprelsa, was the first drug (April 2011) to be approved by US Food and Drug Administration (FDA) for treatment of late-stage (metastatic) medullary thyroid cancer in adult patients who are ineligible for surgery. [13] Cabozantinib , trade name Cometriq, was granted marketing approval (November 2012) by the U.S. ... Retrieved 29 November 2012 . ^ a b Numbers from National Cancer Database in the US, from Page 10 in: F. Grünwald; Biersack, H.
MTC may present as a sporadic form, and in about 30% of cases, as a familial form as a part of multiple endocrine neoplasia (see this term); a dominant inherited disease related to germline mutation of the protooncogene RET .
In families with autosomal dominant inheritance, affected individuals have a 50% chance of passing on the pathogenic variant to offspring. ... COL11A1 may be tested first in individuals with typical ocular findings including type 2 "beaded" congenital vitreous anomaly and significant hearing loss. ... Sequence analysis of the gene of interest is performed first, followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found. ... Systemic abnormalities are not observed. The first signs usually become apparent during early adolescence, but onset can be as early as age two years. VCAN -related vitreoretinopathy is inherited in an autosomal dominant manner. High-grade myopia is a refractive error greater than or equal to −6 diopters.
A number sign (#) is used with this entry because of evidence that Stickler syndrome type IV (STL4) is caused by homozygous mutation in the COL9A1 gene (120210) on chromosome 6q13. For a general phenotypic description and a discussion of genetic heterogeneity of Stickler syndrome, see 108300. Clinical Features Van Camp et al. (2006) described a consanguineous Moroccan family in which 4 of 10 sibs had features characteristic of Stickler syndrome, including moderate to severe sensorineural hearing loss, moderate to high myopia with vitreoretinopathy, and epiphyseal dysplasia. Nikopoulos et al. (2011) reported 2 sisters in a Turkish family and 1 boy in a Moroccan family with features of autosomal recessive Stickler syndrome. All 3 individuals had myopia, vitreous changes, sensorineural hearing loss, and epiphyseal dysplasia.
Overview Stickler syndrome is a genetic disorder that can cause serious vision, hearing and joint problems. Also known as hereditary progressive arthro-ophthalmopathy, Stickler syndrome is usually diagnosed during infancy or childhood. Children who have Stickler syndrome often have distinctive facial features — prominent eyes, a small nose with a scooped-out facial appearance and a receding chin. They are often born with an opening in the roof of the mouth (cleft palate). While there is no cure for Stickler syndrome, treatments can help control symptoms and prevent complications.
A number sign (#) is used with this entry because of evidence that Stickler syndrome type V (STL5) is caused by homozygous mutation in the COL9A2 gene (120260) on chromosome 1p34. One such family has been reported. For a general phenotypic description and a discussion of genetic heterogeneity of Stickler syndrome, see 108300. Clinical Features Baker et al. (2011) studied a large 5-generation consanguineous pedigree of Asian Indian origin segregating autosomal recessive Stickler syndrome. Affected family members had high myopia, vitreoretinal degeneration, retinal detachment, and mild to moderate sensorineural hearing loss. None of the family members was known to have cleft palate, and although there was short stature in childhood, adult height was thought to be appropriate for this family.
Please help to improve this article by introducing more precise citations. ( December 2012 ) ( Learn how and when to remove this template message ) Stickler syndrome (hereditary progressive arthro-ophthalmopathy) Stickler syndrome is inherited in an autosomal dominant pattern. Specialty Medical genetics Stickler syndrome (hereditary progressive arthro-ophthalmodystrophy) is a group of very rare genetic disorders affecting connective tissue , specifically collagen . [1] Stickler syndrome is a subtype of collagenopathy, types II and XI . Stickler syndrome is characterized by distinctive facial abnormalities, ocular problems, hearing loss, and joint and skeletal problems. It was first studied and characterized by Gunnar B. ... It is a sex independent autosomal dominant trait meaning a person with the syndrome has a 50% chance of passing it on to each child.
Molecular Genetics Kakiuchi et al. (2003) found an association between a -116G polymorphism in the promoter region of the XBP1 gene (194355.0001) and susceptibility to bipolar disorder (odds ratio = 4.6) in Japanese patients.