In 2008, the World Health Organization recognized this lymphoma as a variant of the diffuse large cell lymphomas. [6] Subsequent to this recognition, numerous studies found this lymphoma to occur in a wide range of tissues besides the oral cavity and in individuals with various other predisposing immunodeficiency conditions. [7] In 2017, this Organization classified PBL as the most common member of a rare subgroup of lymphomas termed lymphoid neoplasms with plasmablastic differentiation. [1] See also [ edit ] Lymphoma Epstein-Barr virus-associated lymphoproliferative diseases References [ edit ] ^ a b c d e f g h i Chen BJ, Chuang SS (March 2020). ... Advances in Anatomic Pathology . 27 (2): 61–74. doi : 10.1097/PAP.0000000000000253 . PMID 31725418 . ^ a b c d e f g h i j k Lopez A, Abrisqueta P (2018). ... PMC 7106118 . PMID 32269975 . ^ a b c d e f g h Bhattacharyya S, Bains APS, Sykes DL, Iverson BR, Sibgatullah R, Kuklani RM (December 2019). ... PMID 31494113 . ^ Tchernonog E, Faurie P, Coppo P, Monjanel H, Bonnet A, Algarte Génin M, Mercier M, Dupuis J, Bijou F, Herbaux C, Delmer A, Fabiani B, Besson C, Le Gouill S, Gyan E, Laurent C, Ghesquieres H, Cartron G (April 2017). "Clinical characteristics and prognostic factors of plasmablastic lymphoma patients: analysis of 135 patients from the LYSA group" . ... Human Pathology . 79 : 18–41. doi : 10.1016/j.humpath.2018.05.020 . PMID 29885408 . ^ Ott G, Rosenwald A, Campo E (2013). "Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification" .
Plasmablastic lymphoma is an aggressive form of non-Hodgkin lymphoma . Although the condition most commonly occurs in the oral cavity, it can be diagnosed in many other parts of the body such as the gastrointestinal tract, lymph nodes, and skin. The exact underlying cause of plasmablastic lymphoma is poorly understood; however, it is often associated with suppression of the immune system (i.e. HIV infection , immunosuppressive therapy). There is currently no standard therapy for plasmablastic lymphoma. Treatment usually consists of chemotherapy with or without radiation therapy and hematopoietic stem cell transplantation .
A rare aggressive B-cell non-Hodgkin lymphoma characterized by neoplastic cells resembling B immunoblasts or plasmablasts with a CD20-negative plasmacytic phenotype. The tumor may occur in the oral cavity, the gastrointestinal tract, or other, predominantly extranodal, sites and is typically associated with immunodeficiency or -suppression. The tumor cells are EBV-positive in most cases. Patients often present with disseminated bone involvement. Paraproteinemia may also be detected. Prognosis is generally poor.
ISSN 0197-3851 . PMID 18634119 . ^ Blaas, H.-G.; Eik‐Nes, S. H.; Kiserud, T.; Hellevik, L. ... ISSN 1469-0705 . ^ Kumar HR, Jester AL, Ladd AP (December 2008). "Impact of omphalocele size on associated conditions". ... PMID 26297675 . ^ Kanagawa SL, Begleiter ML, Ostlie DJ, Holcomb G, Drake W, Butler MG (March 2002). ... PMC 3979308 . PMID 21238647 . ^ a b c d e f g Aspelund G, Langer JC (2006). "Abdominal wall defects". ... Surgery . 25 (7): 295–297. doi : 10.1016/j.mpsur.2007.05.014 . ^ a b c d e f g h i Wilson RD, Johnson MP (2004).
A number sign (#) is used with this entry because of evidence that isolated omphalocele can be caused by duplication of genes on chromosome 1p31. Description An omphalocele is an abdominal wall defect limited to an open umbilical ring, and is characterized by the herniation of membrane-covered internal organs into the open base of the umbilical cord. Omphalocele is distinguished from gastroschisis (230750), in which the abdominal wall defect is located laterally to a normally closed umbilical ring with herniation of organs that are uncovered by membranes (summary by Bugge, 2010). On the basis of clinical manifestations, epidemiologic characteristics, and the presence of additional malformations, Yang et al. (1992) concluded that omphalocele and gastroschisis are casually and pathogenetically distinct abdominal wall defects. Omphalocele can be a feature of genetic disorders, such as Beckwith-Wiedemann syndrome (130650) and the Shprintzen-Goldberg syndrome (182210).
The most common complications for both techniques are superficial wound infections, recurrence of the hernia [17] and some people experience pain from the surgical site. [18] See also [ edit ] Fetal development Umbilicoplasty Paraumbilical hernia Omphalocoele References [ edit ] ^ Lissauer T, Clayden G (2007). Illustrated Textbook of Paediatrics (3rd ed.). ... PMID 24554114 . ^ Dalenbäck J, Andersson C, Ribokas D, Rimbäck G (August 2013). "Long-term follow-up after elective adult umbilical hernia repair: low recurrence rates also after non-mesh repairs".
A rare, non-syndromic, abdominal wall malformation characterized by a hernia of the abdominal wall, centered on the umbilical cord, in which the protruding viscera are protected by a sac. Epidemiology In Europe, the prevalence at birth of omphalocele (isolated and syndromic forms combined) is estimated at about 1/8,000 births. At least half of patients have other abnormalities associated. Clinical description Omphalocele occurs very early in development and is typically detected prenatally. The hernia occurs at the umbilical ring allowing protrusion of the abdominal organs (midgut, liver, spleen and gonads). The protruding viscera is encased in a sac consisting of three layers: the outer amniotic layer, a middle layer of Wharton's jelly and the inner peritoneal layer.
Clinical Features An omphalocele is an abdominal wall defect limited to an open umbilical ring, and is characterized by the herniation of membrane-covered internal organs into the open base of the umbilical cord. Omphalocele is distinguished from gastroschisis (230750), in which the abdominal wall defect is located laterally to a normally closed umbilical ring with herniation of organs that are uncovered by membranes (summary by Bugge, 2010). Also see 164750 for an autosomal form. Inheritance Havalad et al. (1979) described a family with 4 affected males in a pedigree pattern suggestive of X-linked inheritance. Two maternal half brothers and 2 grandsons of 1 of them, through a daughter, were affected. Abdomen - Omphalocele Inheritance - X-linked ▲ Close
It has been estimated there are 1.6 million people in the US with post-radiation intestinal dysfunction, a greater number than those with inflammatory bowel disease such as Crohn's disease or ulcerative colitis . [1] Research [ edit ] New agents have been identified in animal studies that may have effects on intestinal radiation injury. [1] The research approach in humans has been reviewed. [14] References [ edit ] ^ a b c d e f g h i j k Hauer-Jensen M, Denham JW, Andreyev HJ (2014). ... PMID 22700443 . ^ Movsas B, Vikram B, Hauer-Jensen M, Moulder JE, Basch E, Brown SL, Kachnic LA, Dicker AP, Coleman CN, Okunieff P (2011). "Decreasing the adverse effects of cancer therapy: National Cancer Institute guidance for the clinical development of radiation injury mitigators" .
Overview Radiation enteritis is inflammation of the intestines that occurs after radiation therapy. Radiation enteritis causes diarrhea, nausea, vomiting and stomach cramps in people receiving radiation aimed at the abdomen, pelvis or rectum. It's most common in people receiving radiation therapy for cancer in the abdomen and pelvic areas. For most people, radiation enteritis is temporary, with inflammation usually subsiding several weeks after treatment ends. But for some, radiation enteritis may continue long after radiation therapy ends or may develop months or years after treatment.
The knee is investigated and found to be normal. [7] The diagnosis requires x-rays of the pelvis, with anteriorposterior (AP) and frog-leg lateral views. [9] The appearance of the head of the femur in relation to the shaft likens that of a "melting ice cream cone", visible with Klein's line . ... Pediatrics . 142 (5): e20181067. doi : 10.1542/peds.2018-1067 . PMID 30348751 . ^ a b c d e f g h i j Kliegman, Robert M. (2011).
Clinical Features Rennie (1967) observed 12 children or adolescents with slipped upper femoral epiphysis and a close relative with the same. Two additional patients had a parent with osteoarthritis of the hip. This disorder was found by Ochsner et al. (1977) in 10 members of a family, with 2 other members showing coxarthrosis which may have been a sequel to subclinical slipped epiphysis. Male-to-male transmission was noted. Hagglund et al. (1986) studied 49 families, performing radiographic examinations of first-degree relatives and interviewing second-degree relatives in 4 of the 49 families. Slipped femoral capital epiphysis was found in 1 or more first-degree relatives. In another 13 families, radiographic signs were found. Three generations were affected in the family reported by Hagglund and Hansson (1986).
Epiphysiolysis of the hip is a rare osteonecrosis disorder characterized by unilateral or bilateral disruption of the capital femoral physis with varying degrees of posterior epiphysis translation and simultaneous anterior metaphysis displacement. Patients typically present in pre-adolescence/adolescence with pain of variable intensity in varying locations (hip, groin, thigh, knee).
The signs and symptoms are divided into one group that can appear after an intake of as little as 100 mg of caffeine (roughly the amount contained in a cup of brewed coffee) and another group of symptoms that appear at higher levels of intake (more than 1 g per day). Low-dose symptoms include restlessness, nervousness, excitement, insomnia, flushed face, diuresis (increased urination), and gastrointestinal disturbance. ... Retrieved 15 January 2014 . ^ Winston AP, Hardwick E, Jaberi N (2005). "Neuropsychiatric effects of caffeine" .
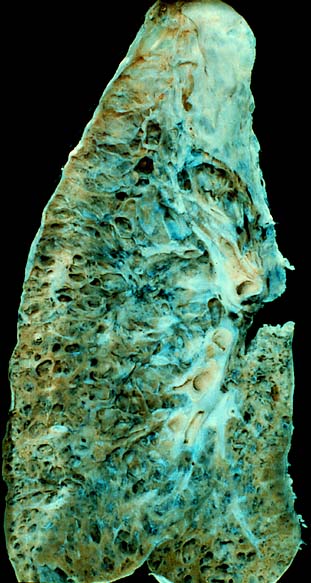
They represent the majority of cases of interstitial lung diseases (up to two-thirds of cases). [8] They were subclassified by the American Thoracic Society in 2002 into 7 subgroups: [ citation needed ] Idiopathic pulmonary fibrosis (IPF): the most common subgroup Desquamative interstitial pneumonia (DIP) Acute interstitial pneumonia (AIP): also known as Hamman-Rich syndrome Nonspecific interstitial pneumonia (NSIP) Respiratory bronchiolitis-associated interstitial lung disease (RB-ILD) Cryptogenic organizing pneumonia (COP): also known as Bronchiolitis Obliterans Organizing Pneumonia (BOOP) Lymphoid interstitial pneumonia (LIP) Secondary [ edit ] Secondary ILDs are those diseases with a known etiology, including: Connective tissue and Autoimmune diseases Sarcoidosis Rheumatoid arthritis Systemic lupus erythematosus Systemic sclerosis Polymyositis Dermatomyositis Antisynthetase syndrome Inhaled substances Inorganic Silicosis Asbestosis Berylliosis Industrial printing chemicals (e.g. carbon black, ink mist) Organic Hypersensitivity pneumonitis (Extrinisic allergic alveolitis) Drug-induced Antibiotics Chemotherapeutic drugs Antiarrhythmic agents Infection Coronavirus disease 2019 [9] Atypical pneumonia Pneumocystis pneumonia (PCP) Tuberculosis Chlamydia trachomatis Respiratory Syncytial Virus Malignancy Lymphangitic carcinomatosis Predominately in children Diffuse developmental disorders Growth abnormalities deficient alveolarisation Infant conditions of undefined cause ILD related to alveolar surfactant region Diagnosis [ edit ] Pneumocystis pneumonia can present with interstitial lung disease, as seen in the reticular markings on this AP chest x-ray . A chest X-ray demonstrating pulmonary fibrosis due to amiodarone Investigation is tailored towards the symptoms and signs. ... Daniele Macchini, Treating COVID-19 Patients in Bergamo, Italy, Describes Horrible Situation" . 11 March 2020. ^ a b c d e f g Ryu JH, Olson EJ, Midthun DE, Swensen SJ (November 2002). ... MayoClinic.com . ^ Hayes D, Jr; Wilson, KC; Krivchenia, K; Hawkins, SMM; Balfour-Lynn, IM; Gozal, D; Panitch, HB; Splaingard, ML; Rhein, LM; Kurland, G; Abman, SH; Hoffman, TM; Carroll, CL; Cataletto, ME; Tumin, D; Oren, E; Martin, RJ; Baker, J; Porta, GR; Kaley, D; Gettys, A; Deterding, RR (1 February 2019). ... PMID 25284270 . ^ Kotloff RM, Thabut G (July 2011). "Lung transplantation".
Overview Interstitial (in-tur-STISH-ul) lung disease describes a large group of disorders, most of which cause progressive scarring of lung tissue. The scarring associated with interstitial lung disease eventually affects your ability to breathe and get enough oxygen into your bloodstream. Interstitial lung disease can be caused by long-term exposure to hazardous materials, such as asbestos. Some types of autoimmune diseases, such as rheumatoid arthritis, also can cause interstitial lung disease. In some cases, however, the causes remain unknown. Once lung scarring occurs, it's generally irreversible.
Surgeon General issued an advisory, and in 1986 the Food and Drug Administration required a Reye syndrome-related warning label for all aspirin-containing medications. [21] References [ edit ] ^ a b c d e f g h i j "NINDS Reye's Syndrome Information Page" . ... Retrieved August 8, 2016 . ^ a b c d e f g h i j k l m n o p q r s t Pugliese, A; Beltramo, T; Torre, D (October 2008). ... PMID 3415311 . ^ Autret-Leca E, Jonville-Béra AP, Llau ME, et al. (2001). "Incidence of Reye's syndrome in France: a hospital-based survey". Journal of Clinical Epidemiology . 54 (8): 857–62. doi : 10.1016/S0895-4356(00)00366-8 . PMID 11470397 . ^ Reye RD, Morgan G, Baral J (1963). "Encephalopathy and fatty degeneration of the viscera.
Overview Reye's syndrome, also known as Reye syndrome, is a rare but serious condition that causes swelling in the liver and brain. Reye's syndrome can occur at any age but usually affects children and teenagers after a viral infection, most commonly the flu or chickenpox. Symptoms such as confusion, seizures and loss of consciousness need emergency treatment. Early diagnosis and treatment of Reye's syndrome can save a child's life. Aspirin has been linked with Reye's syndrome, so use caution when giving aspirin to children or teenagers for fever or pain.
A rare, systemic disease characterized by persistent vomiting with confusion, lethargy, disorientation, hyperreflexia, hyperventilation, and tachycardia, with rapid progression to seizures, non-inflammatory encephalopathy, coma and death. It typically develops between 12 hours and 3 weeks after recovery from a viral illness, such as upper respiratory tract infection or gastroenteritis. Hepatomegaly, acute hepatic steatosis, fatty liver degeneration and multiple laboratory abnormalities are associated.
S2CID 22850257 – via 1573-6709. ^ Desmond Morris, The Naked Ape Trilogy (1994) p. 279-80 ^ Sigmund Freud, On Sexuality (PFL 7) p. 68n ^ Quoted in O. ... Journal of Pediatric Psychology . 4 (4): 406. doi : 10.1093/jpepsy/4.4.403 . ^ Giannini, AJ; Colapietro, G; Slaby, AE; Melemis, SM; Bowman, RK (1998). ... Loeb Classical Library, Harvard University Press ^ Levin, Daniel B. (2005) EPATON BAMA ('Her Lovely Footstep'): The Erotics of Feet in Ancient Greece . ^ Coulton, G. G. (1923) Life in the Middle Ages Volume 3: Men and Manners Chapter 30 - Tricks of Trade .
It is unknown why males are predominantly affected, with rates in males being 17- to 70-fold those in females, despite thyroid overactivity being much more common in women. [1] [2] History [ edit ] Carl Friedrich Otto Westphal After several case reports in the 18th and 19th centuries, periodic paralysis was first described in full by the German neurologist Carl Friedrich Otto Westphal (1833–1890) in 1885. [7] [8] In 1926 the Japanese physician Tetsushiro Shinosaki, from Fukuoka , observed the high rate of thyroid disease in Japanese people with periodic paralysis. [9] [10] The first English-language report, in 1931, originated from Dunlap and Kepler, physicians at the Mayo Clinic ; they described the condition in a patient with features of Graves' disease . [2] [10] In 1937 periodic paralysis was linked with hypokalemia, as well as precipitation of attacks with glucose and insulin. [11] [12] This phenomenon has been used as a diagnostic test. [12] In 1974 it was discovered that propranolol could prevent attacks. [13] The concept of channelopathies and the link with specific ion channel mutations emerged at the end of the 20th century. [1] [3] [4] References [ edit ] ^ a b c d e f g h i j k l m n o p q r s t u v w x y Kung AW (July 2006). ... The Journal of Clinical Endocrinology and Metabolism . 91 (7): 2490–5. doi : 10.1210/jc.2006-0356 . PMID 16608889 . ^ a b c d e f g h i j k Pothiwala P, Levine SN (2010). ... ISBN 978-0-12-374527-9 . PMID 19185183 . ^ a b c d e f g h Lin SH (January 2005). "Thyrotoxic periodic paralysis" (PDF) . Mayo Clinic Proceedings . 80 (1): 99–105. doi : 10.4065/80.1.99 . PMID 15667036 . ^ a b c d Weetman AP (October 2000). "Graves' disease".
PMC 6940034 . PMID 31921379 . ^ a b c d e f g h Sukswai N, Lyapichev K, Khoury JD, Medeiros LJ (November 2019). ... Pathology . doi : 10.1016/j.pathol.2019.08.013 . PMID 31735345 . ^ a b c d e f g h i j k Korkolopoulou P, Vassilakopoulos T, Milionis V, Ioannou M (July 2016). ... "Expression patterns of the activator protein-1 (AP-1) family members in lymphoid neoplasms".
T-cell/histiocyte rich large B cell lymphoma (THRLBCL) is a rare variant of diffuse large B-cell lymphoma (DLBCL; see this term), mainly affecting middle-aged men and often not being discovered until an advanced disease stage, with involvement of the spleen, liver and bone marrow occurring at a greater frequency than in DLBCL. It is often difficult to diagnose due to its similarity with other lymphoid diseases such as classic Hodgkin lymphoma and nodular lymphocyte-predominant Hodgkin lymphoma (see these terms) and has an aggressive clinical course.
., in critical illness polyneuropathy supportive and preventive therapy are important for the affected individual, as well as, avoiding (or limiting) corticosteroids . [22] See also [ edit ] Hereditary neuropathy with liability to pressure palsy Mononeuropathy Neuritis Neuromuscular disease Neuromuscular medicine Neuropathy Polyradiculoneuropathy Radial neuropathy References [ edit ] ^ a b c d e f g h i "Polyneuropathies. Medical information about polyneuropathy | Patient" . ... PMC 4032209 . PMID 24696504 . ^ a b c d e f g h "Polyneuropathy/differential diagnosis" . ... External links [ edit ] Classification D ICD - 10 : G60 - G64 ICD - 9-CM : 356.4 , 357.1 - 357.7 MeSH : D011115 External resources Patient UK : Polyneuropathy Scholia has a topic profile for Polyneuropathy . v t e Diseases relating to the peripheral nervous system Mononeuropathy Arm median nerve Carpal tunnel syndrome Ape hand deformity ulnar nerve Ulnar nerve entrapment Froment's sign Ulnar tunnel syndrome Ulnar claw radial nerve Radial neuropathy Wrist drop Cheiralgia paresthetica long thoracic nerve Winged scapula Backpack palsy Leg lateral cutaneous nerve of thigh Meralgia paraesthetica tibial nerve Tarsal tunnel syndrome plantar nerve Morton's neuroma superior gluteal nerve Trendelenburg's sign sciatic nerve Piriformis syndrome Cranial nerves See Template:Cranial nerve disease Polyneuropathy and Polyradiculoneuropathy HMSN Charcot–Marie–Tooth disease Dejerine–Sottas disease Refsum's disease Hereditary spastic paraplegia Hereditary neuropathy with liability to pressure palsy Familial amyloid neuropathy Autoimmune and demyelinating disease Guillain–Barré syndrome Chronic inflammatory demyelinating polyneuropathy Radiculopathy and plexopathy Brachial plexus injury Thoracic outlet syndrome Phantom limb Other Alcoholic polyneuropathy Other General Complex regional pain syndrome Mononeuritis multiplex Peripheral neuropathy Neuralgia Nerve compression syndrome v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
Lenhoff et al. (2001) evaluated 5 patients with Williams syndrome for absolute musical pitch (AP; see 159300), which is the ability to recognize, name, and reproduce the pitch of a musical note without reference. ... By comparison, cognitively intact musicians who claim to have AP scored 84.3% on similar tests. Lenhoff et al. (2001) suggested that the prevalence of AP in individuals with Williams syndrome is higher than that in the general Western population (1 in 10,000) and noted that the age window of AP acquisition in Williams syndrome appears to be extended compared to the general population.
Williams syndrome is a genetic condition that affects many parts of the body. Signs and symptoms include mild to moderate intellectual disability; unique personality traits; distinctive facial features; and heart and blood vessel problems. Williams syndrome is caused by a person missing more than 25 genes from a specific area of chromosome 7 (a "deletion"). The loss of these genes contributes to the characteristic features. Although Williams syndrome is an autosomal dominant condition, most cases are not inherited and occur sporadically in people with no family history of Williams syndrome. Treatments are based on each person's signs and symptoms, as there is no cure at this time.
Williams syndrome is a developmental disorder that affects many parts of the body. This condition is characterized by mild to moderate intellectual disability or learning problems, unique personality characteristics, distinctive facial features, and heart and blood vessel (cardiovascular) problems. People with Williams syndrome typically have difficulty with visual-spatial tasks such as drawing and assembling puzzles, but they tend to do well on tasks that involve spoken language, music, and learning by repetition (rote memorization). Affected individuals have outgoing, engaging personalities and tend to take an extreme interest in other people. Attention deficit disorder (ADD), problems with anxiety, and phobias are common among people with this disorder.
Note: WS cannot be identified by routine analysis of G-banded chromosomes or other conventional cytogenetic banding techniques. ... Feeding difficulties leading to failure to gain weight are common, including gastroesophageal (G-E) reflux, disordered suck and swallow, textural aversion, and vomiting. Prolonged colic (>4 months) may be related to G-E reflux, chronic constipation, and/or idiopathic hypercalcemia. ... Chronic abdominal pain is a common complaint of children and adults with WS; possible causes include G-E reflux, hiatal hernia, peptic ulcer disease, cholelithiasis, diverticulitis, ischemic bowel disease, chronic constipation, and somatization of anxiety. ... The treatment of feeding problems in infancy and abdominal pain in children and adults depends on the cause (e.g., G-E reflux, hypercalcemia, hiatal hernia, and/or diverticulitis).
GeneReviews . PMID 20301427 . ^ a b c d e f g h i j Reference, Genetics Home (December 2014). ... PMID 11770962 . ^ Sacks, O.; Schlaug, G.; Jancke, L.; Huang, Y.; Steinmetz, H. (1995). ... S2CID 39114788 . ^ Van Strien, J. W.; Lagers-Van Haselen, G. C.; Van Hagen, J. M.; De Coo, I. ... S2CID 24853662 . ^ Kapp, M. E.; von Noorden, G. K.; Jenkins, R (1995). "Strabismus in Williams syndrome". ... PMID 19255058 . ^ Van Der Geest, J. N.; Lagers-Van Haselen, G. C.; Van Hagen, J. M.; Brenner, E.; Govaerts, L.
. ^ Pregnancy-related pelvic girdle pain (PPP), I: Terminology, clinical presentation, and prevalence European Spine Journal Vol 13, No. 7 / Nov. 2004 W. H. Wu, O. G. Meijer, K. Uegaki, J. M. A. Mens, J. ... Östgaard. ^ Villar J, Say L, Gulmezoglu AM, Meraldi M, Lindheimer MD, Betran AP, Piaggio G; Eclampsia and pre-eclampsia: a health problem for 2000 years. ... S2CID 24784165 . ^ Reyes H, Sandoval L, Wainstein A, Ribalta J, Donoso S, Smok G, Rosenberg H, Meneses M (January 1994). ... Oxford University Press. pp. 1587–601. ^ a b c Kourtis AP, Read JS, Jamieson DJ (June 2014).
A number sign (#) is used with this entry because of evidence that Wiedemann-Steiner syndrome (WDSTS) is caused by heterozygous mutation in the MLL gene (KMT2A; 159555) on chromosome 11q23. The KMT2A gene is involved in histone modification. Description Wiedemann-Steiner syndrome is a congenital malformation syndrome characterized by hypertrichosis cubiti associated with short stature; consistent facial features, including long eyelashes, thick or arched eyebrows with a lateral flare, wide nasal bridge, and downslanting and vertically narrow palpebral fissures; mild to moderate intellectual disability; behavioral difficulties; and hypertrichosis on the back (summary by Jones et al., 2012 and Miyake et al., 2016). Clinical Features Wiedemann et al. (1989) reported a Caucasian boy with pre- and postnatal growth deficiency, psychomotor delay, and a round and flat face, short nose, hypertelorism, long philtrum, short palpebral fissures, low-set ears, and high-arched palate. Other findings included an alternating convergent squint, dilatation of the renal calyces, and short and thick limbs. MacDermot et al. (1989) reported a mother and daughter with hairy elbows who also had short stature, rhizomelic shortening of the limbs, round face, and heavy jaw.
Description The KMT2A gene, or MLL, encodes a DNA-binding protein that methylates histone H3 (see 602810) lys4 (H3K4) and positively regulates expression of target genes, including multiple HOX genes (see 142980). MLL is a frequent target for recurrent translocations in acute leukemias that may be characterized as acute myeloid leukemia (AML; 601626), acute lymphoblastic leukemia (ALL), or mixed lineage (biphenotypic) leukemia (MLL). Leukemias with translocations involving MLL possess unique clinical and biologic characteristics and are often associated with poor prognosis. MLL rearrangements are found in more than 70% of infant leukemias, whether the immunophenotype is more consistent with ALL or AML6, but are less frequent in leukemias from older children. MLL translocations are also found in approximately 10% of AMLs in adults, as well as in therapy-related leukemias, most often characterized as AML, that develop in patients previously treated with topoisomerase II inhibitors for other malignancies.
Wiedemann-Steiner syndrome is a rare, genetic multiple congenital anomalies/dysmorphic syndrome characterized by short stature, hypertrichosis cubiti, facial dysmorphism (hypertelorism, long eyelashes, thick eyebrows, downslanted, vertically narrow, long palpebral fissures, wide nasal bridge, broad nasal tip, long philtrum), developmental delay, and mild to moderate intellectual disability. It has a variable clinical phenotype with additional manifestations reported including muscular hypotonia, patent ductus arteriosus, small hands and feet, hypertrichosis on the back, behavioral difficulties, and seizures.
Investigation [ edit ] Two views should be obtained: AP and lateral. PA radiography will look very similar to a Colles' fracture, with a fracture along the distal metaphysis of the radius (can be shortened or comminuted).
This article is about the vertebral fracture. For the wrist fracture, see Smith's fracture . A Smith fracture is a named vertebral fracture occurring most commonly in the lumbar spine . It is similar to that of a Chance fracture and is associated with seat belt injuries. This fracture represents a fracture through the posterior elements including the superior articular processes but not the spinous process, as well as an avulsion fracture of the vertebral body. [1] It is not to be confused with the more commonly referred to Smith's fracture of the wrist. Notes [ edit ] ^ Richard H. Daffner - Chance-Type Fractures of the Thoracolumbar Spine: Imaging Analysis: Discussion References [ edit ] Smith NS, Kaufer H: Patterns and mechanisms of lumbar injuries associated with lapseat belts.
Mishalany et al. (1970) described 2 children with duodenal atresia, all 4 parents of whom were descendants from one couple related as first cousins. Mishalany et al. (1971) then reported that a third affected child had been born in this kindred. Der Kaloustian et al. (1974) reported yet another affected child. See jejunal atresia (243600). Best et al. (1989) described 2 families: in 1, a son and daughter of unrelated parents were affected; in the second, 2 sons of a consanguineous marriage were affected as well as the daughter of 1 of the affected males. In a 10-year survey of 65 hospitals by the surgical section of the American Academy of Pediatrics, 503 patients were identified with congenital duodenal atresia; 13 instances of intestinal atresia in sibs were found, with 4 sibs affected in 1 family (Fonkalsrud et al., 1969).
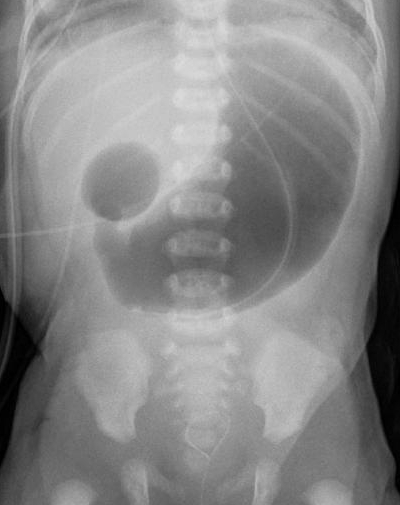
Duodenal atresia is an embryopathy of the cranial intestine that leads to a complete absence of the duodenal lumen. Epidemiology The incidence of duodenal atresia is between 1/10,000 and 1/6,000 live births, with an approximately equal male to female ratio. Clinical description In 30-52% of infants it is an isolated anomaly, but it is often associated with other congenital abnormalities. Approximately 20 to 30% of infants with duodenal atresia are carriers of trisomy 21, and about 20 to 25% have cardiac anomalies. Other frequently described associated malformations include duodenal growth failure, annular pancreas (see this term), which are particular clinical forms of duodenal atresia, and anomalies of the biliopancreatic tract or choledochal cysts.