-
Testicular Dysgenesis Syndrome
Wikipedia
PMID 11331648 . ^ VIRTANEN, H; RAJPERTDEMEYTS, E; MAIN, K; SKAKKEBAEK, N; TOPPARI, J (1 September 2005). ... PMID 16005920 . ^ Toppari, J; Larsen, J C; Christiansen, P; Giwercman, A; Grandjean, P; Guillette, L J; Jégou, B; Jensen, T K; Jouannet, P (1996-08-01). "Male reproductive health and environmental xenoestrogens" . ... PMID 8880001 . ^ Skakkebæk, N. E.; Meyts, E. Rajpert-De; Main, K. M. (2001-05-01). "Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects: Opinion" . ... PMID 37351 . ^ Skakkebæk, N. E.; Meyts, E. Rajpert-De; Main, K. M. (2001-05-01). "Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects: Opinion" . ... PMID 26582516 . ^ Skakkebæk, N. E.; Meyts, E. Rajpert-De; Main, K. M. (2001-05-01). "Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects: Opinion" .
-
Fanconi Syndrome
Wikipedia
Fanconi syndrome Specialty Nephrology , endocrinology Fanconi syndrome or Fanconi's syndrome ( English: / f ɑː n ˈ k oʊ n i / , / f æ n -/ ) is a syndrome of inadequate reabsorption in the proximal renal tubules [1] of the kidney . ... See also [ edit ] Familial renal disease in animals for Fanconi syndrome in Basenjis References [ edit ] ^ " Fanconi syndrome " at Dorland's Medical Dictionary ^ Fanconi Syndrome at Merck Manual Home Health Handbook ^ Magen D, Berger L, Coady MJ, Ilivitzki A, Militianu D, Tieder M, Selig S, Lapointe JY, Zelikovic I, Skorecki K (March 2010). "A loss-of-function mutation in NaPi-IIa and renal Fanconi's syndrome". ... PMID 17494094 . ^ Assmann N, Dettmer K, Simbuerger JM, Broeker C, Nuernberger N, Renner K, Courtneidge H, Klootwijk ED, Duerkop A, Hall A, Kleta R, Oefner PJ, Reichold M, Reinders J (May 2016). ... PMID 15722646 . ^ Hashimoto T, Arakawa K, Ohta Y, Suehiro T, Uesugi N, Nakayama M, Tsuchihashi T (2007). ... External links [ edit ] Classification D ICD - 10 : E72.0 ICD - 9-CM : 270.0 MeSH : D005198 DiseasesDB : 11687 SNOMED CT : 40488004 External resources MedlinePlus : 000333 eMedicine : ped/756 v t e Inborn error of amino acid metabolism K → acetyl-CoA Lysine /straight chain Glutaric acidemia type 1 type 2 Hyperlysinemia Pipecolic acidemia Saccharopinuria Leucine 3-hydroxy-3-methylglutaryl-CoA lyase deficiency 3-Methylcrotonyl-CoA carboxylase deficiency 3-Methylglutaconic aciduria 1 Isovaleric acidemia Maple syrup urine disease Tryptophan Hypertryptophanemia G G→ pyruvate → citrate Glycine D-Glyceric acidemia Glutathione synthetase deficiency Sarcosinemia Glycine → Creatine : GAMT deficiency Glycine encephalopathy G→ glutamate → α-ketoglutarate Histidine Carnosinemia Histidinemia Urocanic aciduria Proline Hyperprolinemia Prolidase deficiency Glutamate / glutamine SSADHD G→ propionyl-CoA → succinyl-CoA Valine Hypervalinemia Isobutyryl-CoA dehydrogenase deficiency Maple syrup urine disease Isoleucine 2-Methylbutyryl-CoA dehydrogenase deficiency Beta-ketothiolase deficiency Maple syrup urine disease Methionine Cystathioninuria Homocystinuria Hypermethioninemia General BC / OA Methylmalonic acidemia Methylmalonyl-CoA mutase deficiency Propionic acidemia G→ fumarate Phenylalanine / tyrosine Phenylketonuria 6-Pyruvoyltetrahydropterin synthase deficiency Tetrahydrobiopterin deficiency Tyrosinemia Alkaptonuria / Ochronosis Tyrosinemia type I Tyrosinemia type II Tyrosinemia type III / Hawkinsinuria Tyrosine → Melanin Albinism : Ocular albinism ( 1 ) Oculocutaneous albinism ( Hermansky–Pudlak syndrome ) Waardenburg syndrome Tyrosine → Norepinephrine Dopamine beta hydroxylase deficiency reverse: Brunner syndrome G→ oxaloacetate Urea cycle / Hyperammonemia ( arginine aspartate ) Argininemia Argininosuccinic aciduria Carbamoyl phosphate synthetase I deficiency Citrullinemia N-Acetylglutamate synthase deficiency Ornithine transcarbamylase deficiency / translocase deficiency Transport / IE of RTT Solute carrier family : Cystinuria Hartnup disease Iminoglycinuria Lysinuric protein intolerance Fanconi syndrome : Oculocerebrorenal syndrome Cystinosis Other 2-Hydroxyglutaric aciduria Aminoacylase 1 deficiency Ethylmalonic encephalopathy Fumarase deficiency Trimethylaminuria v t e Kidney disease Glomerular disease See Template:Glomerular disease Tubules Renal tubular acidosis proximal distal Acute tubular necrosis Genetic Fanconi syndrome Bartter syndrome Gitelman syndrome Liddle's syndrome Interstitium Interstitial nephritis Pyelonephritis Balkan endemic nephropathy Vascular Renal artery stenosis Renal ischemia Hypertensive nephropathy Renovascular hypertension Renal cortical necrosis General syndromes Nephritis Nephrosis Renal failure Acute renal failure Chronic kidney disease Uremia Other Analgesic nephropathy Renal osteodystrophy Nephroptosis Abderhalden–Kaufmann–Lignac syndrome Diabetes insipidus Nephrogenic Renal papilla Renal papillary necrosis Major calyx / pelvis Hydronephrosis Pyonephrosis Reflux nephropathy
-
Cherry Eye
Wikipedia
See also [ edit ] Conjunctivitis , commonly referred to as pink eye References [ edit ] ^ a b c d e f g h Gelatt, K. N. (2000). Essentials of Veterinary Ophthalmology . Baltimore: Lippincott Williams & Wilkins. ^ a b c d e f Gelatt, K. N. (2001). Color Atlas of Veterinary Ophthalmology . Baltimore: Lippincott Williams & Wilkins. ^ a b c d e f g h i j k l m n o Slatter, D. (2001). Fundamentals of Veterinary Ophthalmology : Third Edition. ... Retrieved 1 December 2012. ^ a b c d e f g h Plummer, C., Kallberg, M., Gelatt, K., Gelatt, J., & Barrie, K. P. (2008).

-
Apraxia Of Lid Opening
Wikipedia
Eur Neurol. 2001. 45(1):53-4 ^ Abe K, Fujimura H, Tatsumi C, Toyooka K, Yorifuji S, Yanagihara T. ... J Neurol Neurosurg Psychiatry. 1995 Dec. 59(6):629-32 ^ Schmidtke K, Büttner-Ennever JA. Nervous control of eyelid function. ... Toxicon. 2008 Dec 6 ^ Yamada S, Matsuo K, Hirayama M, Sobue G. The effects of levodopa on apraxia of lid opening: A case report. ... Mov Disord. 1994 Jan. 9(1):116-7 ^ Kerty E, Eidal K. Apraxia of eyelid opening: clinical features and therapy.
-
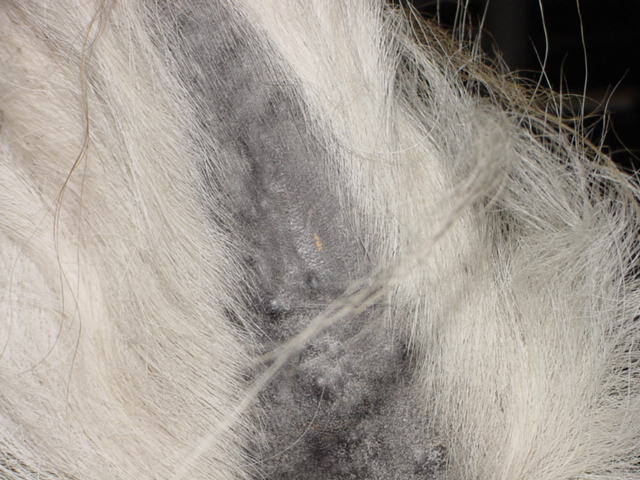
Equine Melanoma
Wikipedia
S; Shaw, E; Buechner‐Maxwell, V; Scarratt, W. K; Crisman, M; Furr, M; Robertson, J (2013). ... PMID 12141308 . ^ a b c Pielberg, G.; Golovko, A.; Sundström, E.; Curik, I.; Lennartsson, J.; Seltenhammer, M.; Druml, T.; Binns, M.; Fitzsimmons, C.; Lindgren, G.; Sandberg, K.; Baumung, R.; Vetterlein, M.; Strömberg, S.; Grabherr, M.; Wade, C.; Lindblad-Toh, K.; Pontén, F.; Heldin, C.; Sölkner, J.; Andersson, L. (2008). ... Journal of the American Veterinary Medical Association . 229 (10): 1617–1622. doi : 10.2460/javma.229.10.1617 . ^ Goetz, T. E.; Ogilvie, G. K.; Keegan, K. G.; Johnson, P. J. (1990).
-
Neointimal Hyperplasia
Wikipedia
. ^ a b c d Serrano, M. Concepcion; Vavra, Ashley K; Jen, Michele; Hogg, Melissa E; Murar, Jozef; Martinez, Janet; Keefer, Larry K; Ameer, Guillermo A; Kibbe, Melina R (2011). ... Circulation . 105 (24): 2917–22. doi : 10.1161/01.cir.0000018168.15904.bb . PMID 12070123 . ^ Shah, P. K (2003). "Inflammation, Neointimal Hyperplasia, and Restenosis: As the Leukocytes Roll, the Arteries Thicken" . ... PMID 10618299 . ^ Kay, I. P; Wardeh, A. J; Kozuma, K; Foley, D. P; Knook, A. H. M; Thury, A; Sianos, G; Van Der Giessen, W.REN, PIK3CA, CCL2, APOE, PIK3CG, PIK3CD, PIK3CB, ISYNA1, FOXO3, NOS2, CDKN1B, NOS3, ADIPOQ, RNF10, IL6, MBD2, NOXA1, HMGB1, SIRT1, GABPA, MMP9, ACTB, NFE2L2, S1PR1, SERPINF1, TRPC1, POLD1, TGFB1, AGT, MAPK3, TAGLN, PCNA, HMOX1, CDK1, MIR21, CETP, PNPLA2, XBP1, MIR222, UCP2, VEGFA, MIR221, VWF, MIR22, NR4A3, MIR29A, PEA15, TNFSF12, CREG1, NRP1, KAT2B, MIR223, MIR320A, ACSS2, SUV39H1, MTCO2P12, TMED7-TICAM2, SELE, SELP, SOS1, STAT3, STIM1, CCR2, MIR33A, SPAG11A, NCF1, PLF, TGM2, TNF, TP53, TNFSF12-TNFSF13, NR1I2, IER3, MIR155, SETD2, SIRT3, CAVIN1, ANGPTL2, PLA2G15, POLDIP2, SERP1, ACCS, AKT1S1, MIR146A, TMED7, CKLF, TNFRSF12A, NOD2, SARAF, DYM, KDM3A, KDM6B, MMRN1, CELIAC5, TICAM2, KLF4, MIR145, MIR126, MFN2, NR1H3, RAD50, SPRY1, SEMA3A, ENTPD8, SPAG11B, CIB1, ZNRD2, DCTN6, NFAT5, S100A12, S100B, PRKG1, REST, EPHB4, DAPK3, DBN1, DCN, DDX5, DOCK2, S1PR3, EFNB2, ELN, EPO, CTSS, EZH2, F10, FHL2, FMOD, GATA4, GATA6, GDNF, GIP, DAPK1, CTSL, PTPRJ, CASP1, AGTR1, AKT1, ANGPT2, BMP2, BMP3, BSG, TSPO, CALCA, CAV1, CTSK, CD36, CD59, CDK4, CDK9, CHGA, CLU, MAPK14, CTNNB1, GJA1, HDAC2, HGF, PRKAA1, NFKB1, NPPC, NUCB2, SERPINE1, PDZK1, PECAM1, PLAU, PPARG, PRKAA2, HIF1A, PRKAB1, MAPK6, PSMD9, PTCH1, PTEN, PTGER2, PTGS2, PTPN1, MYO1E, MYH11, MYBPH, MYO1F, HMGB2, IFI27, IFI35, IFNG, CCN1, IL1B, IRF1, JAK3, KCNN4, LEP, LGALS1, MARCKS, SMAD3, MFAP4, MMP14, MSRA, COX2, H3P23
-
Mixed-Phenotype Acute Leukemia
Wikipedia
Jude Children's Research Hospital , Tennessee, the name "acute leukaemia with mixed lymphoid and myeloid phenotype" was introduced. [14] The World Health Organization in its WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues (2008) adopted the name "mixed-phenotype acute leukemia" to include leukemias of ambiguous lineage, acute undifferentiated leukemias and natural killer lymphoblastic leukemias. [4] References [ edit ] ^ a b Weinberg, O K; Arber, D A (2010). "Mixed-phenotype acute leukemia: historical overview and a new definition" . ... PMID 19570749 . ^ Khan, M; Siddiqi, R; Naqvi, K (2018). "An update on classification, genetics, and clinical approach to mixed phenotype acute leukemia (MPAL)". ... PMC 4896164 . PMID 27233483 . ^ Slany, R. K. (2009-07-01). "The molecular biology of mixed lineage leukemia" . ... PMID 30438955 . ^ Prentice, A. G.; Smith, A. G.; Bradstock, K. F. (1980). "Mixed lymphoblastic-myelomonoblastic leukemia in treated Hodgkin's disease" .
-
Factor X Deficiency
Wikipedia
Produced in the liver FX when activated cleaves prothrombin to generate thrombin in the intrinsic pathway of coagulation. This process is vitamin K dependent and enhanced by activated factor V . ... In the acquired form of FX deficiency an insufficient amount of factor X is produced by the liver due to liver disease, vitamin K deficiency , buildup of abnormal proteins in organs ( amyloidosis ) or certain medications (i.e. warfarin ). [1] In amyloidosis FX deficiency develops as FX and other coagulation factors are absorbed by amyloid fibrils. [3] Diagnosis [ edit ] Blood tests are needed to differentiate FX deficiency from other bleeding disorders. [1] Typical are normal thrombin time , prolonged prothrombin time (PT) and prolonged partial thromboplastin time (PTT). [1] FX antigen and its coagulant activity can be used to classify the severity of the condition: [4] Type I has low levels of FX antigen and activity. ... While effective this treatment carries a risk of blood-borne viruses and fluid overload. If vitamin K levels are low, vitamin K can be supplied orally or parenterally.

-
Aceruloplasminemia
Wikipedia
Pagon RA; Adam MP; Ardinger HH; Bird TD; Dolan CR; Fong CT; Smith RJH; Stephens K (eds.). "Aceruloplasminemia" . GeneReviews . PMID 20301666 . Retrieved 2014-02-12 . ^ Yoshida, K; Furihata, K; Takeda, S; Nakamura, A; Yamamoto, K; Morita, H; Hiyamuta, S; Ikeda, S; Shimizu, N; Yanagisawa, N (March 1995).

-
Mismatch Repair Cancer Syndrome
Wikipedia
It was first reported by Canadian surgeon Jacques Turcot (1914-1977 ) et al. in 1959 and hence carries the first author's name. [9] See also [ edit ] Gardner syndrome References [ edit ] ^ a b c d Online Mendelian Inheritance in Man (OMIM): 276300 ^ Kratz CP, Holter S, Etzler J, Lauten M, Pollett A, Niemeyer CM, Gallinger S, Wimmer K (June 2009). "Rhabdomyosarcoma in patients with constitutional mismatch-repair-deficiency syndrome" (PDF) . ... PMID 19293170 . S2CID 42347878 . ^ Wimmer K, Etzler J (September 2008). "Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg?". ... S2CID 32654505 . ^ Krüger S, Kinzel M, Walldorf C, Gottschling S, Bier A, Tinschert S, von Stackelberg A, Henn W, Görgens H, Boue S, Kölble K, Büttner R, Schackert HK (January 2008). ... PMID 18376293 . ^ Jackson CC, Holter S, Pollett A, Clendenning M, Chou S, Senter L, Ramphal R, Gallinger S, Boycott K (June 2008). "Café-au-lait macules and pediatric malignancy caused by biallelic mutations in the DNA mismatch repair (MMR) gene PMS2".PMS2, MLH1, MSH6, MSH2, APC, FBXO11, TP53, CD274, MRC1, BRAF, KRAS, ARID1A, ERBB2, PTEN, CDX2, EGFR, MBD4, CTNNB1, PIK3CA, TYMS, POLE, MSH3, MGMT, IGF2R, PDCD1, BAX, TGFBR2, FAP, PTGS2, SELENOP, TGFB1, SOX9, SMARCB1, ALCAM, MDC1, CCNO, CHEK2, DICER1, MLH3, FOXP3, CHFR, PINK1, PRRT2, GBP5, MCM9, MIR148A, MIR155, IKZF1, NF1, PMS1, PLK1, AR, BAAT, BRCA1, BRCA2, CASP3, CASP7, CD80, CDK4, CDKN2A, ERCC1, ESR1, FANCB, HIF1A, HRAS, EPCAM, MHS6, MAP3K11, MUC2, MUC5AC, MUC6, ALDOB, NF2, OPA1, PAX5, PIK3CB, MIR224
-
Thumb Hypoplasia
Wikipedia
Therefore, a careful examination of both hands is important. [3] Treatment [ edit ] When it comes to treatment it is important to differentiate a thumb that needs stability, more web width and function, or a thumb that needs to be replaced by the index finger. [4] Severe thumb hypoplasia is best treated by pollicization of the index finger. [3] [5] Less severe thumb hypoplasia can be reconstructed by first web space release, ligament reconstruction and muscle or tendon transfer. [3] [5] It has been recommended that pollicization is performed before 12 months, but a long-term study of pollicizations performed between the age of 9 months and 16 years showed no differences in function related to age at operation. [3] It is important to know that every reconstruction of the thumb never gives a normal thumb, because there is always a decline of function. [4] When a child has a good index finger, wrist and fore-arm the maximum strength of the thumb will be 50% after surgery in comparison with a normal thumb. [3] [4] The less developed the index finger, wrist and fore-arm is, the less strength the reconstructed thumb will have after surgery. [3] [4] References [ edit ] ^ a b c d e f g h i j k l m n o p q r Riley, S.A. & Burgess, R.C. (2009). ... Journal of Hand Surgery, vol 34A, 1564–1573 ^ a b c d e f g h i j k l Ashbaugh, H. & Gellman, H. (2009). ... Journal of Craniofacial Surgery, vol 20, number 4, 1039–1044 ^ a b c d e f g h i j k l m n o Manske, P.R. & Goldfarb, C.A. (2009). ... London, United Kingdom: Informa Healthcare ^ a b c d e f g h i j k l m n Light, T.R. & Gaffey, J.L. (2010).RPS19, RPL11, MIR17HG, ESCO2, TFAP2A, NSD2, NELFA, GDF5, RECQL4, SEMA3E, EIF4A3, KDM1A, SIN3A, WIPI2, APC, CHST11, BCOR, CHD7, SALL4, PALB2, EHMT1, ADAMTS10, LMNB2, UBE2T, ACAN, RPL5, FLNA, ATP6V1B2, BMPR1B, COL2A1, DHCR7, DHODH, FANCA, FANCC, FANCD2, FANCE, FBN1, HOXA13, IHH, LETM1, LRP4, LTBP2, MGP, PTCH1, RAD51C, BHLHA9, RASA1, CUP2Q35, RGS6
-
Hypotrichosis With Juvenile Macular Dystrophy
Wikipedia
Journal of Investigative Dermatology . 132 (10): 2332–41. doi : 10.1038/jid.2012.171 . PMID 22696062 . Nagel-Wolfrum, K; Möller, F; Penner, I; Wolfrum, U (2014). ... Gregory-Evans, C. Y.; Wang, X; Wasan, K. M.; Zhao, J; Metcalfe, A. L.; Gregory-Evans, K (2014). ... N.; Kanuga, N.; Wolfrum, U.; Nagel-Wolfrum, K.; Da Cruz, L.; Coffey, P. J.; Cheetham, M.

-
Multisystemic Smooth Muscle Dysfunction Syndrome
Wikipedia
.; Davies, R.; Haan, E. A.; Holman, K. J.; Watson, K. C.; Sreetharan, D.; Cao, S. ... PMID 10532176 . ^ Milewicz, Dianna M.; Østergaard, John R.; Ala-Kokko, Leena M.; Khan, Nadia; Grange, Dorothy K.; Mendoza-Londono, Roberto; Bradley, Timothy J.; Olney, Ann Haskins; Adès, Lesley; Maher, Joseph F.; Guo, Dongchuan; Buja, L.
-
Polyphagia
Wikipedia
PMID 21499159 . ^ Masuzaki H, Tanaka T, Ebihara K, Hosoda K, Nakao K (2009). "Hypothalamic melanocortin signaling and leptin resistance--perspective of therapeutic application for obesity-diabetes syndrome" .
-
Quadricuspid Aortic Valve
Wikipedia
., Passeri, J., Tighe, D., & Agnihotri, A. K. (2011). Quadricuspid aortic valve: a report of 12 cases and a review of the literature. ... G., Yang, H. S., Lee, D. H., Shin, J. K., Chee, H. K., & Kim, J. S. (2014).
-
Equine Multinodular Pulmonary Fibrosis
Wikipedia
. ^ a b c d Marenzoni ML, Passamonti F, Lepri E, Cercone M, Capomaccio S, Cappelli K, et al. (July 2011). "Quantification of Equid herpesvirus 5 DNA in clinical and necropsy specimens collected from a horse with equine multinodular pulmonary fibrosis" . ... PMID 21908328 . ^ a b c Back H, Kendall A, Grandón R, Ullman K, Treiberg-Berndtsson L, Ståhl K, Pringle J (September 2012).
-
Young–simpson Syndrome
Wikipedia
Young–Simpson syndrome Other names Hypothyroidism-dysmorphism-postaxial polydactyly-intellectual disability syndrome, Say-Barber-Biesecker-Young-Simpson syndrome This condition is inherited via autosomal dominant manner Young–Simpson syndrome ( YSS ) is a rare congenital disorder with symptoms including hypothyroidism , heart defects , facial dysmorphism , cryptorchidism in males, hypotonia , mental retardation and postnatal growth retardation . [1] [2] Other symptoms include transient hypothyroidism, macular degeneration and torticollis . [3] The condition was discovered in 1987 and the name arose from the individuals who first reported the syndrome. [4] [5] An individual with YSS has been identified with having symptoms to a similar syndrome known as Ohdo Blepharophimosis syndrome , showing that it is quite difficult to diagnose the correct condition based on the symptoms present. [6] Some doctors therefore consider these syndromes to be the same. [7] The mode of inheritance has had mixed findings based on studies undertaken. [5] [8] One study showed that the parents of an individual with YSS are unrelated and phenotypically normal, indicating a sporadic mutation , thus making it difficult to base the cause of the condition on genetic makeup alone. [5] However, another study was done of an individual with YSS who had first cousins as parents, giving the possibility of autosomal recessive inheritance. [8] Contents 1 KAT6B 2 See also 3 References 4 External links KAT6B [ edit ] In 2011, it was demonstrated that de novo mutations in the gene KAT6B caused YSS. [9] See also [ edit ] Genitopatellar syndrome References [ edit ] ^ Masuno M, Imaizumi K, Okada T, et al. (May 1999). "Young-Simpson syndrome: further delineation of a distinct syndrome with congenital hypothyroidism, congenital heart defects, facial dysmorphism, and mental retardation". ... PMID 10602125 . ^ Young ID, Simpson K (November 1987). "Unknown syndrome: abnormal facies, congenital heart defects, hypothyroidism, and severe retardation" . ... PMID 8474111 . ^ Clayton-Smith, Jill; O'Sullivan James; Daly Sarah; Bhaskar Sanjeev; Day Ruth; Anderson Beverley; Voss Anne K; Thomas Tim; Biesecker Leslie G; Smith Philip; Fryer Alan; Chandler Kate E; Kerr Bronwyn; Tassabehji May; Lynch Sally-Ann; Krajewska-Walasek Malgorzata; McKee Shane; Smith Janine; Sweeney Elizabeth; Mansour Sahar; Mohammed Shehla; Donnai Dian; Black Graeme (November 2011).

-
Uterine Clear-Cell Carcinoma
Wikipedia
PMID 18637491 . ^ a b c C A Hamilton; M K Cheung; K Osann; L Chen; N N Teng; T A Longacre; M A Powell; M R Hendrickson; D S Kapp & J K Chan (Mar 2006).
-
Testicular Atrophy
Wikipedia
ISSN 0309-0167 . PMID 3371900 . ^ Pinto, K. J.; Kroovand, R. L.; Jarow, J. P. ... PMID 2891525 . ^ Gray, T. J.; Butterworth, K. R. (1980). "Testicular atrophy produced by phthalate esters". ... PMID 6776936 . ^ Uraki, Ryuta; Hwang, Jesse; Jurado, Kellie Ann; Householder, Sarah; Yockey, Laura J.; Hastings, Andrew K.; Homer, Robert J.; Iwasaki, Akiko; Fikrig, Erol (2017-02-22).
-
Pulmonary Sclerosing Pneumocytoma
Wikipedia
Respir Med 107(3):448-50. doi: 10.1016/j.rmed.2012.12.005 ^ Soo IX, Sittampalam K, Lim CH (2017) Pulmonary sclerosing pneumocytoma with mediastinal lymph node metastasis. ... Jpn J Clin Oncol 16(1):77-86 ^ Yamazaki K (2004) Type-II pneumocyte differentiation in pulmonary sclerosing hemangioma: ultrastructural differentiation and immunohistochemical distribution of lineage-specific transcription factors (TTF-1, HNF-3 alpha, and HNF-3 beta) and surfactant proteins. ... Cytojournal 13:9. doi: 10.4103/1742-6413.180783. ^ Hara K, Izumi N, Tsukioka T, Komatsu H, Okada S, Toda M, Ito R, Shibata T, Nishiyama N (2016) Multiple pulmonary sclerosing pneumocytoma with abnormal accumulation of fluorodeoxyglucose-positron emission tomography diagnosed by durgical treatment; Report of a case.NKX2-1, AKT1, AR, BRAF, SMUG1, TP53, CTNNB1, ARHGEF2, TTF1, TUBA1A, TP63, ACTB, RABEPK, STK11, EBNA1BP2, RASSF1, ARMH1, LANCL1, S100A1, SFTPB, S100B, PTEN, PSMD7, MAPK1, MMP9, RPSA, KRAS, IL9, ESR2, CEACAM5, CDKN2A, CD34, H3P28







