-
Glycogen Storage Disease Type Vi
GeneReviews
If present, hypoglycemia is mild and may manifest during an illness after prolonged fasting. Ketotic hypoglycemia after an overnight fast is the salient feature of this disorder. ... Disorders to Consider in the Differential Diagnosis of Glycogen Storage Disease Type VI View in own window Disorder Gene(s) MOI Features of Differential Diagnosis Disorder Overlapping w/GSD VI Distinguishing from GSD VI Phosphorylase kinase deficiency (GSD IX) PHKA2 PHKB PHKG2 XL AR Hepatomegaly Fasting ketosis Hypoglycemia ↑ AST/ALT ↑ lipids Male predominance AST & ALT commonly more severely ↑ Hepatic glycogen synthase deficiency (GSD 0) (OMIM 240600) GYS2 AR Fasting hypoglycemia Ketosis Absence of hepatomegaly Postprandial hyperglycemia & hyperlactatemia Glucose-6-phosphatase deficiency (GSD Ia) G6PC1 AR Hepatomegaly Fasting hypoglycemia ↑ AST/ALT Hyperlipidemia Severe fasting lactic acidosis Hyperuricemia Marked hyperlipidemia Glucose-6-phosphate transporter deficiency (GSD Ib) SLC37A4 AR Hepatomegaly Fasting hypoglycemia ↑ AST/ALT Hyperlipidemia Neutropenia Crohn’s disease Hyperuricemia Debranching enzyme deficiency (GSD III) AGL AR Hepatomegaly Fasting hypoglycemia ↑ AST/ALT Hyperlipidemia Low prealbumin AST & ALT usually markedly ↑ Muscle involvement w/↑ CK Branching enzyme deficiency (GSD IV) GBE1 AR Hepatomegaly ↑ AST/ALT ↓ prealbumin Lack of hypoglycemia until end-stage liver disease GLUT2 deficiency (Fanconi-Bickel syndrome; GSD XI) (OMIM 227810) SLC2A2 AR Hepatomegaly Fasting hypoglycemia Fasting ketosis ↑ AST/ALT Low prealbumin Postprandial hyperglycemia Chronic diarrhea Hypophosphatemic rickets Fanconi nephropathy Fructose-1,6-bisphosphatase deficiency 1 FBP1 AR Hepatomegaly Fasting hypoglycemia ↑ AST/ALT Fasting hyperlactatemia Alpha-1 antitrypsin deficiency-related hepatitis 2 SERPINA1 AR Hepatomegaly ↑ AST/ALT Lack of fasting hypoglycemia & ketosis Glycerol kinase deficiency (OMIM 307030) GK XL Hypoglycemia Ketoacidosis & extremely ↑ glycerol PRKAG2 deficiency (see Hypertrophic Cardiomyopathy Overview) PRKAG2 AD Nonlysosomal glycogen accumulation primarily in skeletal & cardiac muscle Ventricular pre-excitation & mild-to-severe cardiac hypertrophy No hypoglycemia Niemann-Pick disease type B 3 (see ASM Deficiency) SMPD1 AR Hepatomegaly Growth failure Hyperlipidemia No fasting hypoglycemia Significant splenomegaly Bone & pulmonary involvement Gaucher disease 3 GBA Adapted from Kishnani et al [2019] AD = autosomal dominant; AR = autosomal recessive; ASM = acid sphingomyelinase; GSD = glycogen storage disease; MOI = mode of inheritance; XL = X-linked 1. ... Treatment of Manifestations in individuals with GSD VI View in own window Manifestations/ Concern Treatment Considerations/Other Hypoglycemia Frequent small meals Uncooked cornstarch (1-1.5 g/kg) 1-4x/day Protein 2-3 g/kg body weight per day Glycosade 1 from waxy maize, proven beneficial in children age >5 yrs & adults to extend overnight fast duration Doses adjusted to keep glucose concentrations at 75-100 mg/dL (4.2-5.6 mmol/L) & beta-OH-butyrate concentrations ≤0.2 mmol/L Hepatomegaly Restricted intake of simple sugars (<5 g) Restricted intake of total carbohydrates (15-30 g per meal) To ↓ liver size Growth restriction Cornstarch & protein supplementation Growth normalizes w/treatment Growth hormone contraindicated Decreased bone density Cornstarch & protein supplementation Calcium & Vitamin D Primarily due to ketosis Muscle cramping Protein 2-3 grams per kg body weight per day Muscle cramping is usually due to undertreatment & protein deficiency. 1. ... This experimental product may improve maintenance of normoglycemia with fasting for a longer duration and may reduce the number of doses of cornstarch required.
-
Hyperacusis
Wikipedia
However, tinnitus is more common [5] and there are important differences between their involved mechanisms. [1] Contents 1 Signs and symptoms 1.1 Associated conditions 2 Causes 3 Neurophysiological mechanisms 4 Diagnosis 5 Treatment 6 Notable cases 7 See also 8 External links 8.1 Information Websites 8.2 Non-profit Organizations 9 References 10 Further reading Signs and symptoms [ edit ] In hyperacusis, the symptoms are ear pain, annoyance , distortions, and general intolerance to many sounds that most people are unaffected by. ... Voice actor Liam O'Brien has hyperacusis, and is quoted as having lost sleep during the time of diagnosis. [33] See also [ edit ] Auditory system Hearing Misophonia Otoacoustic emission Photophobia Tinnitus Tinnitus masker External links [ edit ] Information Websites [ edit ] A 2020 study by the NIHR Nottingham Biomedical Research Centre analyzed the content and quality of information of fifteen popular hyperacusis websites using the validated DISCERN questionnaire. [34] The website Hyperacusis Focus achieved the highest overall DISCERN score. The reviewers found that Hyperacusis Focus and U.K. National Health Service websites were the most comprehensive online resources for health care professionals and patients, respectively. ... Content and Quality of Hyperacusis Information Websites [35] Website DISCERN Score Hyperacusis Focus 4.13 Action on Hearing Loss 3.41 American Speech-Language-Hearing Association 3.28 Dizziness & Balance 3.19 National Health Service 3.00 Wikipedia 2.69 British Tinnitus Association 2.34 WebMD 2.34 Hyperacusis.net 2.22 Non-profit Organizations [ edit ] Hyperacusis Research Limited is a 501(c)(3) non-profit charity dedicated to the development of effective treatments for hyperacusis and to funding research which will eliminate the underlying mechanisms that cause hyperacusis.CREBBP, EDNRA, TBL2, GTF2IRD1, BAZ1B, CLIP2, TNF, SLC1A3, RFC2, OTX2, LIMK1, GTF2I, ESR1, EP300, ELN, MLXIPL, ERBB2, SCN11A

-
Body Mass Index Quantitative Trait Locus 18
OMIM
For a phenotypic description and a discussion of genetic heterogeneity of body mass index (BMI), see 606641. Molecular Genetics Based on the interaction of MRAP2 with MC4R (155541), an energy homeostasis protein implicated in obesity, and the phenotype of Mrap2-null mice, Asai et al. (2013) investigated whether alterations in MRAP2 are associated with human obesity.
-
Hemolytic Anemia
Wikipedia
The heme is ultimately converted to bilirubin and removed in stool and urine. [38] Hemoglobin may be cleared directly by the kidneys resulting in fast clearance of free hemoglobin but causing the continued loss of hemosiderin loaded renal tubular cells for many days. ... Am Fam Physician. 2004;69:2599–606. [PubMed] [Google Scholar] ^ a b Stanley L Schrier, MD. ... Retrieved 6 November 2013 . External link in |website= ( help ) ^ Mary Anna Thrall, Dale C.G6PD, TPI1, SLC4A1, GCLC, COL4A1, HMOX1, ITPA, SPTB, GPI, EPO, IFNA2, GSR, EIF2AK1, ALAS2, A2M, UROS, ADAMTS13, NT5C3A, SPTA1, HBA2, HBA1, PGK1, CD59, ANK1, GSS, KRAS, CD40LG, RHAG, SLC2A1, HBB, PTPN22, TNFRSF13B, ATP7B, KCNN4, ATP11C, TNFRSF13C, LCAT, NFKB1, NFKB2, BTNL2, NRAS, PFKM, PGM3, PIGA, PRKCD, PIGT, ICOS, ALAD, HLA-DRB1, AK1, KDM6A, WAS, WIPF1, KMT2D, LAT, TNFSF12, TREX1, UROD, CD81, GP1BA, FCGR2B, CTLA4, FECH, GALT, DNASE1, CR2, GATA1, EPB42, CD19, MS4A1, EPB41, FCGR2A, CFH, PKLR, KLF1, HFE, UGT1A1, ADA, PRDX2, CP, HAMP, RN7SL263P, UBL4A, DHS, PIEZO1, H6PD, GCLM, ABCG8, HEPH, MIR451A, MIR144, GSTK1, BTG3, ADD2, IFNL3, IL27, SLEH1, SMUG1, C20orf194, SIRPA, MYDGF, ATN1, SLCO6A1, CRYZ, IL37, HPGDS, DECR1, KRT88P, FOXP3, ACR, RAB14, CD55, MED25, IL17D, ECB2, FUBP1, MTHFR, PTGS2, PSEN1, FOLR1, FOLR2, CD47, ABO, PRDX1, COX2, RPS19, CD247, C3, TNFRSF17, HP, BRCA2, HBG2, HK1, FLNA, S100A8, DMTN, UGT1A, SMARCA5, CDR3, F2, F3, FANCD2, VWF, FBP1, UGCG, SLC6A3, SLC35A2, TRV-AAC1-4, FCGR3A, TFRC, TRIM21, ALDH9A1, ALDOA, MTCO2P12
-
Brachydactyly Type C
GARD
Brachydactyly type C is a very rare congenital condition that is characterized by shortening of certain bones in the index, middle and little fingers. The bones of the ring finger are typically normal. Other abnormalities may also be present such as hypersegmentation (extra bones) of the index and middle fingers; ulnar deviation (angled towards the fifth finger) of the index finger; and unusually-shaped bones and/or epiphysis (end of a long bone).
-
Non-Communicable Disease
Wikipedia
A trend has emerged, particularly in the early 2000s, in which numerous studies have revealed a link between fast food and an increase in heart disease. These studies include those conducted by the Ryan Mackey Memorial Research Institute, Harvard University and the Sydney Center for Cardiovascular Health. Many major fast food chains, particularly McDonald's, have protested the methods used in these studies and have responded with healthier menu options. ... CHRODIS: EU Joint Action on Chronic Diseases and Promoting Healthy Ageing Across the Life-Cycle WHO website on non-communicable diseases WHO Regional Office for the Eastern Mediterranean website on non-communicable diseases "NCDnet — Global Noncommunicable Disease Network" .

-
Diabetes In Cats
Wikipedia
These symptoms arise from the body's inability to use glucose as an energy source. A fasting glucose blood test will normally be suggestive of diabetes at this point. ... In combination with dehydration, fasting, infection, or other body stresses, the condition may progress to diabetic ketoacidosis , a medical emergency with a high fatality rate that cannot be treated at home. ... The human synthetic insulin, Humulin N /Novolin N/ NPH, is usually a poor choice for cats, [16] since cats metabolize insulin about twice as fast. The Lente and Ultralente versions were popular for feline use until summer 2005, when they were discontinued. ... Caninsulin (known in the US as Vetsulin) is a brand of porcine-based insulin approved for cats which is available with a veterinarian's prescription. According to the manufacturer's website, the insulin's action profile in cats was similar to that of NPH insulin, and it lowered blood sugar quickly, but for only about 6–8 hours. ... Some veterinarians still use the outdated recommendation of using Humulin "N" or NPH insulin for cats, which is very fast-acting for most cats. [14] The slower-acting Lente and Ultralente (Humulin L and Humulin U) insulins were discontinued in 2005), so most cats are treated with either the veterinary PZI insulins or the new full-day analogs glargine (Lantus) and detemir (Levemir).
-
Patent Ductus Arteriosus (Pda)
Mayo Clinic
Sweating with crying or eating. Persistent fast breathing or breathlessness. Easy tiring. ... This quick and simple test records the electrical signals that make up the heartbeat. It shows how fast or how slowly the heart is beating. ... Are there brochures or other printed material I can have? What websites do you recommend? Don't hesitate to ask other questions, as well.
-
Gastroparesis
Mayo Clinic
Tests may include: Gastric emptying tests To see how fast your stomach empties its contents, one or more of these tests may be recommended: Scintigraphy. ... Samples of your breath are collected over a few hours and the amount of the substance in your breath is measured. The test can show how fast your stomach empties after consuming food by measuring the amount of the substance in your breath. ... Are there brochures or other printed material that I can take with me? What websites do you recommend? Do I need a follow-up visit?HMOX1, POLG, TRNL1, HTRA2, COX1, COX2, COX3, ND1, ND4, ND5, ND6, TRNF, TRNH, CLASP1, RRM2B, TRNS1, TRNS2, TRNW, PARK7, POLG2, PRKN, PODXL, DNAJC6, SNCA, SON, TRNQ, UCHL1, VPS13C, SPATA5, ATP7A, PINK1, IRF2BPL, TYMP, SELENON, TWNK, SLC25A4, TMEM70, RNU4ATAC, LRRK2, GCG, GHSR, GABPA, MLN, NFE2L2, ALB, SYMPK, MGAM, GPR55, GOLGA6A, CCR9, NPNT, EHMT1, CD160, MLIP, RETNLB, B3GAT1, SOX6, RCBTB1, ANO1, TNF, PART1, ABL2, NOS1, TAC1, SRF, AMBP, APOE, FAS, ARG1, STS, RERE, CCK, CHAT, CSF1, DUSP1, FAAH, FOXF1, FOXF2, GFAP, GH1, GLP1R, HCRTR1, HTR4, IL1B, IL6, KIT, MRC1, OPRM1, PAEP, PDGFRA, MAPK8, PRL, SCN10A, SI, AK6
-
Dilated Cardiomyopathy
Mayo Clinic
Signs and symptoms of dilated cardiomyopathy may include: Fatigue Shortness of breath (dyspnea) during activity or while lying down Reduced ability to exercise Swelling (edema) in the legs, ankles, feet or belly (abdomen) Chest pain or discomfort Fast, fluttering or pounding heartbeat (palpitations) When to see a doctor If you are short of breath or have other symptoms of dilated cardiomyopathy, see your health care provider as soon as possible. ... An electrocardiogram (ECG) can show how fast or how slow the heart is beating. ... Are there brochures or other printed materials I can have? What websites do you recommend? What to expect from your doctor Your provider is likely to ask you several questions, including: Do you always have symptoms or do they come and go?TTN, SCN5A, DMD, ACTC1, TNNT2, MYH6, TMPO, ADRB1, NPPA, ADRB2, SOD2, FAS, LMNA, EGFR, MYH7, TNNI3, PSEN1, ABCC9, RAF1, SGCD, BAG3, ACTA1, SDHA, ALMS1, PSEN2, DES, CSRP3, LDB3, CAP2, GPX1, CTNNB1, NKX2-5, ITGB1, WDR12, SLC22A5, CD36, CAVIN4, NR3C2, PKP2, NPPB, UCP1, TNF, AXIN2, TCF7L2, SHBG, SGCB, RAC1, CSF3, RENBP, FASN, FASLG, SIK1, CSRNP1, ATM, ABRA, AGT, TP53, PDLIM5, LRRC10, AGTR1, SRF, TBX20, BCL2, DAG1, MRGPRD, PTGER4, DNM1L, ILK, MED1, ESRRB, ERBB2, PPARGC1A, MYOCD, GNAQ, CTSV, BAX, TUBB, PPP3CA, NR2F2, PROX1, MAS1, TIMP3, TMOD1, CXCR3, RXRA, RPS6KB1, CXCL1, PTPN11, TPM1, RAB1A, COX7A1, BCL2L1, AGTR2, YME1L1, MDM4, PDCD1, CASP8, CASP9, CASP3, IL10, CENPF, PHC1, PLN, SNAI1, LMOD2, DICER1, TAZ, TNNC1, VCL, RBM20, MYBPC3, DNAJC19, MYPN, DSP, ANKRD1, TNNI3K, EYA4, FKTN, FKRP, MMP1, CRYAB, TXNRD2, ACTN2, SYNE1, DSG2, SLC25A4, LAMP2, PRDM16, HSPB7, DPM3, SPEG, DOLK, TMEM43, PPCS, POLG, XK, NEBL, NEXN, TAF1A, TCAP, HADHA, HCG22, PGM1, TRNV, SDHD, SDHAF1, MYL2, TRNW, NDUFB8, CEP85L, RYR1, TTN-AS1, MYZAP, NDUFS2, BOLA3, DSG2-AS1, HAMP, SKI, SLC12A2, PPP1R13L, POLG2, SYNE2, KAT6B, NDUFAF3, ACAD8, ACAD9, ANKRD11, NCAPH2, POMT2, RRM2B, MAP3K20, NDUFB11, LIMS2, TWNK, SELENON, EPG5, TRNK, RBCK1, HAND2-AS1, ZBTB17, HJV, GCOM1, SURF1, TERT, ADSS1, TPM2, TPM3, GATAD1, TSFM, MGME1, DHX16, KCNAB2, HACD1, HAND2, SCO2, SLC2A10, TRNL1, ACADVL, COX7B, ITGA7, HADHB, HADH, COL7A1, HCCS, CHKB, HMGCL, RUNX1, JUP, GLB1, KCNE1, ATP5F1D, RERE, MIPEP, LAMA3, LAMA4, LAMB3, CPT2, LAMC2, ADD2, ND1, ACTB, ND6, ATP6, GABRD, ND5, ND4, ADCY5, ND3, ND2, FHL2, ACE, CXADR, MARCKSL1, EMD, MUC2, FLNC, LGALS3, ZASP, IGF1, HAND1, VEGFA, HLA-DQB1, CASZ1, EDN1, PIK3CA, PIK3CB, SPP1, PIK3CD, MIR208B, FXN, SLC2A4, CA8, NAMPT, OBSCN, GATA4, HSPB6, NFKB1, MINDY3, GFAP, TBX5, TLR4, TEAD1, TGFB1, TGM2, C20orf181, ADIPOQ, EDNRB, PIK3CG, MEF2A, EDNRA, GRK2, RYR2, MMP9, LPL, HLA-DRB1, PPARA, COMP, RNF111, MIR499A, MIR22, TAX1BP3, KLHL24, MIR214, CRMP1, CNN1, MIR25, MIR340, IL22, MIR33A, IL17D, ADORA1, TLR7, TNFRSF12A, CXADRP1, MYOZ2, TRPV2, UFM1, AKT1, DLL1, CCN2, ITGB1BP2, SYNM, E2F6, UFL1, ATN1, DNTT, DAPK2, MICA, PRPF6, ADCYAP1, TOR1AIP1, HAVCR1, CTLA4, CYP2E1, GATA6-AS1, CTSL, PDLIM3, SGSM3, MIR92B, CCR4, CTSB, RGCC, CTNNA3, CHD7, RTN4, MIR21, RBM45, PPP1R2C, DDR1, TLN2, HOPX, ATP5MD, BRCA1, BMI1, BDH1, FBXO32, AXL, LRG1, ATP6V1G2, MTPN, CHRM2, ZKSCAN4, GRK3, ARR3, RND3, AR, AHSG, CBLL2, DNAJB1P1, SLC25A5, ANO5, RBM24, IL27, OPA3, CAD, ARC, CALR, MIR185, ERBIN, MYDGF, BTNL2, MIR18A, MIR148A, CDKN1B, ALDH2, JPH2, CDH2, ALPK3, MYH7B, CD40LG, CD40, SCARB2, NIF3L1, ELAC2, CD34, IFIH1, MIR146A, SLC25A5P8, ADRA2C, CASR, CAMP, MUL1, MYH14, TRNI, PLEKHM2, NCOA6, LTBP2, LPP, LMO7, LHCGR, LDHA, REN, REST, S100A1, SCN1A, LAMA2, CCL2, CXCL12, KDR, KCNN3, SGCA, JARID2, ITPA, SLC6A2, SLC8A1, ISL1, ISG20, IL18, SPG7, IL17A, STAT3, IL7R, TAC1, ADAM17, IL6, MC4R, PRKCE, PRKCA, NRAP, TRNP, MYBPC1, MYBPC2, MTM1, MYLK, NCAM1, NDUFAB1, COX1, NOS1, NOS2, NOS3, NOTCH4, MMP14, TNFRSF11B, PRKAR1A, OSM, SERPINE1, PRKN, PCYT1A, MMP8, PDK1, PDK4, MMP3, ABCB1, MME, MFGE8, PPARG, MDH1, IL5, IL4, IL2RA, IL18BP, DENR, BECN1, TNFSF10, IER3, TIMELESS, NOL3, DNAJA3, GJA5, GJA1, GH1, DNAJC6, ELMO1, NR1I3, TRIM13, ELP1, GAB1, YAP1, FN1, FOXD4, POSTN, CELF1, F3, ESR1, NMU, WWP2, EEF1A2, NLRP1, MMRN1, GRK5, GSN, TIMP1, HRH2, TIMP2, TIMP4, TJP1, TLN1, TLR3, IFNB1, DNAJB1, HSPD1, TNFRSF1B, HSP90AA1, HSPA4, HSPA1B, PRMT1, HRC, LTBP4, HPRT1, TUFM, HLA-G, UTRN, VASP, WNT5A, HGF, HFE, CXCR4, SLMAP, PLA2G7, ARHGEF5, FXR1, GCH1
-
Symphalangism, Distal
OMIM
In the published x-rays, the fusion was most complete in the index finger. Steinberg and Reynolds (1948) provided follow-up on the family of Inman (1924). Cole (1935) described fused distal interphalangeal joint of the index finger only, behaving apparently as an irregular dominant. ... Poush (1988, 1991) observed a multigenerational family with this trait. The index finger was predominantly affected, and the toes were also affected in most subjects. ... Prepubertal x-rays showed a nonossified space and no distal phalanx growth plate in the index finger. One individual with severe involvement of the distal interphalangeal joints was born with craniosynostosis involving the sagittal suture and craniofacial asymmetry.
-
Primary Progressive Aphasia
Mayo Clinic
What you can do When you make the appointment, ask if there's anything you need to do in advance, such as fasting before having a specific test. ... Are there brochures or other printed material I can have? What websites do you recommend? Should I consider genetic testing?GRN, TARDBP, C9orf72, APOE, LAMC2, MAPT, CSF2, NEFL, SMUG1, IGFALS, ECD, PRNP, ACHE, SOD1, TOMM40, TNFRSF10B, PSIP1, KHSRP, VCP, RIDA, ABAT, TNFRSF1B, SIGLEC7, BBC3, TREM2, IL33, FOXP2, LRRK2, BPIFA2, TP53, PTPN4, TCF3, TCF4, ALOX5, APOC1, BDNF, CAT, CHI3L1, CUX1, GBA, HCLS1, IL2RB, MSMB, PPA1, PSPN, PSEN1, PSPH, PTEN, REG1A, STXBP3, AOS

-
Antiphospholipid Syndrome
Mayo Clinic
The most common are heparin and warfarin (Jantoven). Heparin is fast-acting and delivered via injections. ... Are there brochures or other printed material I can have? What websites do you recommend? Don't hesitate to ask other questions, as well.APOH, PPARG, FRMD4A, TSHR, F5, F3, SYCP2L, F2, PTPRO, GPI, ANXA5, TLR4, SH2B2, KLK3, ANXA2, TNF, AGER, CPB2, MTOR, MTHFR, F10, PLG, HT, TFPI, PLAT, SELPLG, ACR, SERPINE1, MOK, LRP8, VWF, HMGB1, VIM, SELP, RAB4A, CCL2, CXCL12, ATXN2, RO60, S100A10, TRIM21, ABCA1, SSB, THBD, TNFRSF1B, NR1I2, ADIPOQ, SH2B3, PROCR, ADAMTS13, TREX1, PTPN22, FOXP3, SLC52A1, IL21, ANXA8, ANXA8L1, PROS1, NOS3, PON1, PLSCR1, HLA-DPB1, GP1BA, GCY, FGA, FCGR2A, F2RL1, EMD, EDN1, DECR1, CRP, CD36, CD1D, CALR, B2M, SERPINC1, AQP4, AMH, HLA-DRB1, HRES1, IDS, MBL2, PF4, PC, SERPINB2, TNFRSF11B, MYD88, MSN, MPL, LPA, IFNG, LGALS9, LCT, CXCL10, CXCL8, IL1B, IGFBP1, IGF1, C20orf181

-
Hirschsprung's Disease
Mayo Clinic
What you can do When you make the appointment, ask if there's anything your child needs to do in advance, such as fasting for a specific test. Make a list of: Your child's signs or symptoms, including details about bowel movements — frequency, consistency, color and associated pain Your child's key medical information, including other conditions he or she has and family medical history All medications, vitamins or supplements your child is taking and how much water he or she drinks in a typical day Questions to ask your child's doctor Take a family member or friend along, if possible, to help you remember the information you're given. ... Are there any brochures or other printed materials I can have? What websites do you recommend? Don't hesitate to ask other questions.EDN3, EDNRB, GDNF, RET, ZEB2, NRG1, SOX10, SEMA3D, SEMA3C, ECE1, GFRA1, NKX2-1, CAVIN2, MIR206, GAL, FZD3, NTRK3, RELN, AFAP1-AS1, WNT3A, MIR369, NRSN1, CELSR3, ARID1B, GAP43, MIR218-1, BMI1, MIR195, MIR128-1, MED12, LCT-AS1, UTP25, IHH, L1CAM, ERBB2, ITGB1, CD14, AEBP2, PHOX2B, PAX3, NRTN, KIFBP, PROKR1, DSCAM, PIGV, ASCL1, BDNF, TCF4, PIGO, RASGEF1A, COMT, RMRP, CSGALNACT2, MITF, KITLG, KIAA0586, RAD51, RPGRIP1L, RIMBP2, PIGN, RAD51C, SETBP1, SH2B1, EXOC6B, TBX1, B9D1, PGAP2, UBE2T, RREB1, SNAI2, KIAA0556, KIR2DS4, KIR2DL1, KIR2DS1, MBTPS2, NPHP1, SEC24C, SF3B4, MAD2L2, PIBF1, PIGL, POLR2F, NAA10, MKKS, XRCC2, GJB6, UFD1, ZNF423, HIRA, KRAS, KIT, KIR3DL1, CEP104, TMEM216, VRK2, TMEM138, CSPP1, AHI1, ARMC9, DDX59, BRIP1, DHCR7, SLX4, ABHD1, CREBBP, PIGY, TMEM67, PGAP3, CEP41, B3GALT6, CEP120, ARX, APLF, ARL13B, CALB2, HYLS1, BRCA2, BRCA1, JMJD1C, MYO1H, ATRX, PIGW, ARVCF, ARL3, LINC00327, ACTG2, TCTN2, CEP290, FANCE, GATA1, FANCF, FANCG, CC2D2A, FOXF1, SALL4, INPP5E, GJB2, PALB2, FANCI, GP1BB, RFWD3, FANCL, MKS1, BCOR, FANCB, SLC6A8, FANCD2, FANCC, TCTN1, TMEM231, CPLANE1, EP300, TMEM237, ERCC4, FANCM, FANCA, ACHE, NTRK1, SLC9A3R2, NRG3, SCAF11, GEMIN2, GFRA4, SLC2A1, SEMA3A, MCS+9.7, BMP4, DNMT3B, FN1, HOXB5, S100A1, NID1, PTCH1, MIR215, PSPN, AKT1, NLGN1, IL11, PROK1, CALCA, MIR141, ICAM1, KCNN3, ANGPTL2, ELP1, PGP, PLAGL2, ARTN, RGS6, BMP2, S100B, MECP2, BMP10, MEG3, GLI1, CX3CL1, VAMP5, MIR483, NTF3, MIR770, ACTR2, AICDA, LRSAM1, SNRNP70, CHRNE, ITIH5, DNMT1, CYP2B6, COL6A1, COL6A2, DECR1, CTH, DCX, DVL2, ITPKC, ESR1, GFRA2, IARS2, GABRG2, ACKR3, FHL1, FGF1, NLGN2, FABP7, ERCC1, DNTT, ENO2, NOX5, EDNRA, DYRK1A, DVL3, DVL1, SVEP1, DUSP6, KCNG4, ZNF827, CDX2, MIR192, MIR214, ADRA2B, MIR24-1, MIR30A, MIR31, ADRA1A, MIR31HG, MIR431, MIR488, COL6A4P2, MIR637, MIR939, C17orf107, MIR1324, HOTTIP, MTRNR2L12, ADARB1, ACTB, FALEC, LINC01844, CBSL, ALK, MIR150, CDX1, MIR146A, TSGA13, CD34, CCK, PROKR2, CBS, GPR42, CAV1, CASR, KCNG3, NLRP6, CAPN1, BRS3, DOK6, BMP5, FGD2, BBS2, ASS1, CCDC66, COL6A4P1, RBPMS2, APP, GLI3, FOXA1, GRB10, WNT8A, CXCR4, MAPK10, DPF3, MAPK3, DVL1P1, PRKCI, FXYD1, CUL3, PLEK, IL1RL2, BANF1, HSPB3, PLAG1, LPAR2, DCLK1, KLF4, PIK3CG, SLIT2, HAND2, ROCK2, PIGA, TUBA1A, WNT1, ANO1, VIP, SCN1B, SCN10A, SIM2, RYR3, RYR2, RYR1, SOX2, SOX4, SOX9, ROCK1, SRY, SSTR4, STC1, SYP, ROBO1, RBP4, TCOF1, RBP3, TTF1, PTPRR, PTGER2, PTGES, PCDH9, PAX6, PCDHA9, IL17RA, BACE2, IL17A, AUTS2, IGF2, IGF1, SIGLEC8, CNTN6, HSPB2, HSPB1, TLX3, KCNK4, HOXA13, MNX1, SUFU, CNTN5, NLGN3, SLC6A20, GSTT1, GSTP1, GSTM1, ITGB2, JAG2, UBR4, CXCR6, EIF4A3, NUP98, ARNT2, NOTCH1, AKT3, NOS2, NOS1, NGF, MT1A, TRAF3IP2, KCNH2, MCC, MAP2, PRDX3, STMN2, INMT, ZNF609, SMAD1, PLCB1, KCNJ12, ABO

-
Chondrosarcoma
Mayo Clinic
It's not often used for chondrosarcoma because this type of cancer often doesn't respond to chemotherapy. But some fast-growing types of chondrosarcoma may respond to this treatment. ... Are there brochures or other printed material that I can take with me? What websites do you recommend? What will determine whether I should plan for follow-up visits?EXT1, COL2A1, MTOR, FGFR3, IDH1, IDH2, NR4A3, EXT2, TAF15, PTH1R, TP53, VEGFA, IL1B, MMP1, BCL2, MMP13, CDKN2A, SOX9, MMP3, HIF1A, RUNX2, CDK4, MMP2, EWSR1, TNF, PIK3CG, MAPK1, HLCS, GLI1, H3P10, ABCB6, VEGFC, CARD14, ACAN, CYCS, CNMD, PRNP, PRDX2, C4BPA, AHSA1, CASP3, EGFR, EDN1, CTNNB1, CCN2, POLDIP2, TNC, MAPK14, CRK, RNF19A, PTHLH, BDNF, STAT3, GRAP2, MMP9, PIK3CD, ABCB1, AREG, AIMP2, PIK3CA, PIK3CB, MET, TGFB1, PPARG, TEC, CXCR4, CD274, GLI2, MDM2, MYC, CCN6, SIRT1, PDCD5, PTGS2, CCL5, BMP2, SIX3, CHN1, TFG, RETN, PEG10, OSCP1, ICAM1, COMP, TIMP1, MIB1, IGF1, DKK1, MMP12, IL1A, RRAD, IL6, CXCL8, IL10, MIR10B, TERT, COX2, AKT1, ITGAV, ACVR2A, ITM2B, ADIPOQ, LDHA, CXCL12, MTCO2P12, SMUG1, PDPN, ETV5, PTCH1, PLAU, TNFSF10, CCND1, PTEN, TNFRSF11A, VHL, MIR30A, FN1, CDK2, SPHK1, MBD2, SOX4, CTSK, CDKN2B, XIAP, ADAMTS16, POSTN, CKAP4, EPHA1, FERMT2, ANGPT1, WIF1, SH2B2, KDM6B, LRCH1, SATB2, SRGAP2, AMBP, HEY1, BRD4, KDM1A, ANXA5, ANGPT2, ADAMTS3, TNFSF11, TP63, BECN1, RIPK1, ADAM15, APRT, CCN4, SOCS3, LATS1, ATG12, SLC9A3R1, AIRE, ADAMTS4, ADAMTS2, NCOA2, BAG3, APOBEC3B, TRAF4, RAB11FIP3, HS3ST1, HDAC6, EDIL3, NAMPT, TOB1, TLR6, TUBA1B, TUBB3, YAP1, AMACR, GDE1, AHSG, HBP1, MIR30B, MIR29B2, MIR29B1, MIR27B, MIR23B, MIR21, MIR206, MIR204, MIR20A, MIR192, MIR186, MIR17, MIR145, MIR100, BCAR4, MIR96, MIR302C, MIR452, MUC5B, CSAG2, COMMD3-BMI1, CD24, HOTAIR, ADAM8, MIR454, MIR624, MIR494, MIR507, MIR519D, MIR518B, MIR525, MIR497, MIR495, CSAG3, CTAG1A, CSAG1, KLK3, ELSPBP1, CIP2A, SLC12A9, SPHK2, BCOR, AGER, HDAC7, MMP28, ZBTB7A, LINC00328, ADIPOR1, AHCY, DCPS, DISC1, PARP1, ADIPOR2, CREBRF, GFM1, GPBAR1, CHDM, MUC16, NAPRT, ADAMTSL1, DNER, SESN2, TET1, ARHGAP24, MAP1LC3B, LBH, SLC2A10, CD276, PDCD1LG2, PPFIBP1, UVRAG, IFT88, MAGEA4, MID1, MFAP1, CCR7, COL1A2, MATN3, MAP2, SMAD1, IL11, LEP, KIT, JUNB, ITGB3, IRF5, INSM1, MIF, MKI67, KMT2A, CCR5, CLU, CLCN3, MMP7, CHEK1, MMP14, MTAP, MTHFR, CDS1, MYOG, NFATC2, CCN3, NRAS, SERPINE1, INHBA, COL4A5, PDGFA, EZH2, GFAP, GDNF, DCN, FGFR2, DRD3, FGFR1, EXT3, IHH, EFNA5, ENG, EPAS1, ESRRB, ESR1, ERBB2, GPC3, GLB1, DAXX, GLS, HDC, HGF, CTAG1B, CSPG4, HMGB1, FOXA1, PRMT1, HSP90AA1, HSPG2, VCAN, COL11A2, IFI16, IGFBP3, PDCD1, PDGFRB, ALX1, TGFB2, TMSB4X, TM7SF2, TLR1, TIMP3, TIMP2, TGFB3, BMPR2, SRC, TFPI, VPS51, PPP1R11, TCF7, TBX3, TAZ, BMP7, BMP6, TP53BP1, TP73, UCP2, UCP3, KDM6A, EPHB2, VCAM1, BMI1, BCL2L1, HEMC, TFEB, TUSC3, ATR, SLC25A16, HMGA2, CA11, SPP1, CDKN1C, PRKAB1, PTH, PTGER1, PROS2P, CD6, MAPK3, CD44, PRKAA2, CASP7, PRKAA1, CD68, PLK1, PLAGL1, CDC25A, CDKN1A, CCNA2, CCK, RUNX3, RAF1, RARG, RB1, RNASE3, RPE65, S100A1, S100B, CCL11, SDC2, SFRP5, SFTPC, SLC12A3, SMARCB1, SMO, ACTB

-
Functional Dyspepsia
Mayo Clinic
What you can do When you make the appointment, ask if there's anything you need to do in advance, such as fasting before having a specific test. ... Are there brochures or other printed material I can have? What websites do you recommend? Don't hesitate to ask other questions.
-
Pinched Nerve
Mayo Clinic
You may need tests to measure your fasting blood glucose or thyroid levels. ... Are there any brochures or other printed material that I can take home with me? What websites do you recommend visiting? In addition to the questions that you've prepared to ask your doctor, don't hesitate to ask other questions during your appointment.
-
Paget's Disease Of Bone
Mayo Clinic
What you can do When you make the appointment, ask if there's anything you need to do in advance, such as fasting before having a specific test. ... Are there brochures or other printed material I can have? What websites do you recommend? Don't hesitate to ask other questions.TNFRSF11A, SQSTM1, OPTN, DCSTAMP, NUP205, PML, CSF1, ZNF687, RIN3, INPP5D, IL6, KHDRBS1, PIGN, NUP62, DCTN4, DPYS, GTF2H1, SLC25A43, VCP, TNFRSF11B, CFDP1, GEMIN4, PSMD2, EIF4G2, PDB1, VDR, DKK1, TNF, ERBB2, ESR1, KRT7, SOST, NFE2L2, IL1A, IL1B, HNRNPA2B1, HNRNPA1, TNFSF11, ATG5, AR, GABPA, CALCA, KEAP1, CEACAM5, PABPN1, SOCS1, LPAR2, GNE, WASHC5, TUBA1B, NEURL1, VAV3, CASP8AP2, ACP5, ATG7, SLCO6A1, AAA1, KRT8P3, STMP1, FSIP2, GSTK1, GOLGA6A, UCMA, C9orf72, ARID2, NFAM1, KMT2C, POSTN, BIRC6, SMURF1, ACKR3, ATG16L1, GDE1, QPCT, TARDBP, TUBA1A, ATF7, CXCR6, AAAS, SPP1, XRCC1, CTNNB1, MTOR, FOS, FKBP5, FKBP4, FGF2, EPHB2, EGF, EDNRA, DNAH8, DMD, CTSB, CSF2, BEST1, CEBPB, CASR, BTF3P11, BRS3, BRCA2, BGLAP, BCL2, APP, APEX1, ADRA2B, ADRA1A, GCG, GJA1, GPR42, GRP, NR1H2, TRIP6, TP53, TIA1, TAF12, TAF2, SSTR4, ACTA1, SOD1, PTH, MAPK1, PDPK1, NFKB2, NEU1, MUC1, KRT8, CXCL8, IL6ST, IL3, IL1R1, IGFALS, IAPP, FOXA1, ELDR
-
Fibroadenoma
Mayo Clinic
But, in some cases, you may need surgery to remove a fast-growing fibroadenoma. When no treatment is needed If results of an imaging test and biopsy show that your breast lump is a fibroadenoma, you may not need surgery to remove it. ... Do you have brochures or other written materials about this topic? What websites do you suggest I use for more information?MED12, RARA, TP53, ESR1, RASSF1, LOC110806263, ERBB2, WNT2, KIT, FST, LMNA, MMP2, MMP11, TP63, HMGA2, DHCR24, PRLR, TERT, UVRAG, CCL2, KHDRBS1, SCGB1D2, IGF2BP1, RB1, RHEB, EDIL3, RPE65, S100A6, CDKL1, ADAM9, VDR, CCL19, SLC5A5, TLR4, NAA10, TMPO, FOSL1, RAB40B, WNT7B, TWIST1, VEGFA, POLD3, ANXA6, CKAP4, ADAMTS8, ACVR1C, SPATA18, ADAMTS18, NUP43, MIR10A, MIR126, MIR183, MIR188, MIR21, MIR221, MIR331, MT1IP, H3P41, H3P30, H3P42, SAT2, SCIN, ADAMTS20, ANAPC7, PTEN, PRAME, CDK20, BRMS1, STEAP1, PELP1, WNT4, CENPK, ALG1, GATAD2B, MTUS1, ZNF398, ZNF410, GOLPH3, PSIP1, POLB, PSMC6, ETS1, DDX5, DNMT1, EGFR, EIF4E, FBL, ESR2, FGF1, DAXX, FGFR4, FKBP4, FLNA, MSTN, GJA1, GTF2H4, DCN, CYP17A1, HIC1, RUNX3, TRIM23, STS, CCND1, BCL2, C1QBP, CASP3, CD74, CYP1A1, CDH1, CDK4, CDKN2A, CCR5, COL5A1, CST6, HDAC2, IARS1, KLK10, MYC, MT1H, MT1JP, MT1M, MT1L, MT1X, MTNR1A, MYD88, MT1F, NF1, NFKB2, PCNA, PGR, AR, PPP1R8, MT1G, MT1E, IFNG, KRT14, IGF2, IL6, CXCR1, IL10, ING1, INHBA, LASP1, MT1B, LTB, MGMT, MMP9, MMP13, MSN, MT1A, H3P10
-
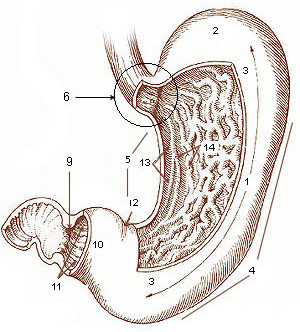
Dumping Syndrome
Mayo Clinic
Causes In dumping syndrome, food and gastric juices from your stomach move to your small intestine in an uncontrolled, abnormally fast manner. This is most often related to changes in your stomach associated with surgery, including any stomach surgery or major esophageal surgery, such as removal of the esophagus (esophagectomy). ... Are there brochures or other printed material that I can take? What websites do you recommend? Don't hesitate to ask other questions during your appointment.