-
Genetic Atypical Hemolytic-Uremic Syndrome
GeneReviews
Surveillance: Serum concentration of hemoglobin, platelet count, and serum concentrations of creatinine, LDH, C3, C4, and haptoglobin: (1) every month in the first year after an aHUS episode, then every three to six months in the following years, particularly for those with normal renal function or chronic renal insufficiency as they are at risk for relapse; and (2) in relatives with the pathogenic variant following exposure to potential triggering events. ... However, the rare individuals with aHUS associated with homozygous pathogenic variants in CFH and very low levels of circulating CFH protein can blur the distinction between HUS and C3G. ... Leukocyte count Other Serum LDH concentration Haptoglobin Serum C3 and C4 concentrations Plasma concentrations of Bb and sC5b-9 Measure serum concentrations of CFH and CFI. ... Measure serum concentration of hemoglobin, platelet count, and serum concentrations of creatinine, LDH, C3, and C4, and haptoglobin: Every month in the first year after an aHUS episode, then every three to six months in the following years, particularly for persons with normal renal function or chronic renal insufficiency as they are at risk for relapse. ... Every two weeks for those rare individuals with homozygous CFH pathogenic variants that result in very low or undetectable levels of the CFH protein Note: The proposed time intervals for checking hemoglobin, platelet count, and serum concentrations of creatinine, LDH, C3, C4, and haptoglobin are suggestions [Authors, personal observation]; each center may follow different guidelines based on their own experience.
-
Lupus Erythematosus
Wikipedia
When a single gene deficiency does cause lupus, it is usually attributed to the complement protein genes C1 , C2 , or C4 . The influence of sex chromosomes and environmental factors are also noteworthy. ... A second-line drug is methotrexate in its low-dose schedule. [14] In 2011, the U.S.CR2, C2, TLR9, IL17A, CD40LG, IL2, IFNA13, DNMT1, FCGR2B, IL10, TRIM21, IL21, TNF, IFNA1, TLR7, IL6, IRF5, TNFSF13B, CD70, TREX1, RO60, MTOR, ESR1, TLR5, ITGAM, IFNG, BTK, IL1B, IL22, CTLA4, FLI1, HLA-DRB1, CD28, BLK, CCL2, FOXP3, TRBV20OR9-2, TP53BP1, MAPK1, EPHB2, FCGR3A, CRP, ICOS, IFIH1, RNPC3, PRL, RAB4A, EZH2, ITGAL, ACE, SPP1, IL2RA, TLR4, MBL2, MECP2, IL4, STAT4, PTPN22, BTG3, CD44, CALR, C3, FCGR3B, KIR3DL2, AIM2, PBX1, C1QA, DECR1, ITGB2, EGFR, CYBB, CREM, KIR3DL1, IL18, IL3RA, IRAK1, F11R, IRF7, ISG20, TLR8, F3, CBLIF, HMGB1, VEGFA, MYDGF, BANK1, HRES1, APOH, STAT3, STAT1, BCL2, CXCL12, BCR, MAVS, IFI16, IFN1@, RFX1, HDAC6, F5, IGHG3, FCGR2A, FLII, MIR146A, MIR21, CASP1, CAMK4, GEM, TREM1, HSPA4, IL1RN, TNFSF4, IFNAR1, KRT20, IL1A, IL23A, IL17D, IRAK4, NR1I3, PRKAR1A, PRKCB, HDAC9, MBD2, REN, TNFSF12, TNFSF13, RPS19, APOL1, SLAMF1, SPG7, SSB, CXCR4, TGFB1, TGFB3, TLR2, VCAM1, PTPA, PPARD, CXCL10, TRIM13, IL21R, KIR2DS1, CD274, LEP, LGALS3, PADI4, SH2D1A, SIRT1, MMP9, MTHFR, MYD88, NCF2, NOS3, P2RX7, PC, CXCL13, PDCD1, LGALS9, TRPM2, FAS, NEAT1, MIR663A, CD69, TNFSF12-TNFSF13, FASLG, FAM167A, AR, UBASH3B, ARR3, CD72, CXADRP1, SLAMF6, MIR155, CXADR, CTNNB1, CAMP, RBM45, EMB, TNFRSF17, CDR1, MIR31, PRDM1, IL27, SRCIN1, CXCR5, MIR148A, STING1, ADAM10, CD19, ACTB, CD38, GATA3, MIR125A, CASR, XRCC6, IGAN1, CD40, VSIR, ETS1, ANXA6, SLEB4, MS4A1, HNRNPDL, TNFSF9, FCGR2C, MIR142, IER3, MIR145, MIR17HG, MIR29B2, MIR29B1, MIR224, ATG12, XPR1, GRAP2, KLK4, MIR23B, TNFSF15, EIF2AK3, ATG5, MIR150, SPATA2, ROCK2, MACROH2A1, CHST12, H4C14, MBTPS1, DGAT1, TAM, AIMP2, SIGLEC14, H3C9P, TMED7-TICAM2, VWF, VIP, RPL17-C18orf32, KLRC4-KLRK1, ERVK-20, VIM, EZR, ERVW-4, VDR, LOC102723971, UMOD, TYK2, H4C9, H4C1, H4C4, H4C15, BECN1, OASL, WG, NR0B2, TAGLN2, LBX1-AS1, H4C13, H4C5, MIR633, H4C2, H4C8, H4C3, H4C11, H4C12, H3C13, H4C6, PSME3, AHSA1, MAEA, WNK1, VTCN1, CD276, SLC38A1, NT5C3A, LBH, TMED7, ELOF1, IL33, DNER, ORMDL3, PRRT2, IL17F, NLRP3, CGAS, CTHRC1, DUOX2, ERVW-1, NAA16, IL25, LRRK2, GORASP1, APOM, IFNK, SPHK2, BDH2, SLC12A9, ABHD6, SCYL1, RIPK4, MRTFA, DUOX1, PHRF1, HAMP, HIVEP3, CD244, RFH1, BACH2, ERVK-6, TBX21, H4-16, TNIP1, PLA2R1, SYNPO, PADI2, RSPO1, CORO1A, LILRA3, LILRB4, H3C15, C1QL1, DCTN6, SH2B2, LARP6, TICAM2, VGLL3, PROCR, ANP32B, ZNRD2, TRIM38, KLRK1, ATF6, H3C14, DKK1, PDCD4, PHGDH, DOCK11, POLDIP2, IL34, IL23R, BTLA, RNF19A, CENPV, TIGIT, TNFRSF13B, BRD4, SGMS1, NBEAL2, KDM6B, ZNF423, IFNL3, NAT2, PROC, TRPC6, TRAF6, F12, F10, F2, ERN1, ERBB2, EPO, EPHX2, ELF1, EIF4EBP1, EGR2, EFNA2, E2F1, DUSP4, TSC22D3, ATN1, ARID3A, DNMT3B, DNMT3A, DNASE1, DLAT, CYP2D6, CYP2B6, CYP1A1, FCER1G, FCN2, FOS, HGF, IFI27, ICAM1, HSPG2, HSPA5, HNRNPC, HMGN2, HLA-H, HLA-DQA1, HLA-C, HIF1A, CFHR1, FOSB, CFH, H1-0, GSTP1, GRIN2A, GRN, GPER1, CXCR3, GPI, GFI1, IFI6, CX3CR1, CTBP2, CSF3R, ATF3, CASP8, CASP6, CAST, C4B, C1R, C1QC, TSPO, BST2, BGN, BCHE, SERPINC1, CAT, KLK3, APOE, ANXA2, ABCD1, AKT1, AGER, PARP1, ADD2, ACP5, ACR, CASP9, RUNX1T1, CSF1, CDKN1A, MAPK14, CRK, CRH, CREBBP, CPD, COX8A, CCR5, CHRM3, AKR1C4, CD52, CDH13, CBL, CDK1, CD48, ENTPD1, CD36, CD80, CD14, CD8B, CD247, CD2, CD1D, IFNAR2, IFNB1, IFNR, PTPRC, CCL3L1, SERPINB3, S100B, RPS6, RPL17, RPA1, TRIM27, RELB, RELA, PTX3, PTPN11, CX3CL1, PTPN6, PTGS2, PTGS1, PSMD9, MASP1, ABCA1, MAP2K1, PRKCD, PRKCA, PPARG, CCL14, SDC2, PIK3CG, SYT1, TRAF2, HSP90B1, TPO, TNFRSF1B, TNFAIP3, TIMP3, THAS, TG, ZEB1, ADAM17, SYK, SELP, SUV39H1, SRI, SNRPB, SNRPA, SNRNP70, SNCA, SIGLEC1, SLC4A1, SH3BP2, SRSF1, PON1, PIK3CD, IL5, KIR2DL4, MFGE8, CD46, MCL1, CD180, LY6E, LDLR, LCT, LCN2, LBR, LAG3, KIF5A, AFF1, JUND, JUNB, JUN, ITGAX, ITGAE, IRF6, TNFRSF9, IL16, IL15, IL9, CIITA, MMP2, PIK3CB, NOTCH1, PIK3CA, SERPINA1, PGK1, PF4, PDE8A, PDE7A, PCNA, OCA2, NT5E, PNP, NHS, MMP11, NFKB1, NF1, NEDD9, NCL, COX1, MSH5, MRC1, MPO, MPL, MNAT1, H3P23

-
Mitochondrial Short-Chain Enoyl-Coa Hydratase 1 Deficiency
GeneReviews
The diagnosis of ECHS1D is established in a proband by the identification of biallelic pathogenic variants in ECHS1 on molecular genetic testing or low short-chain enoyl-CoA hydratase (SCEH) activity using cultured skin fibroblasts. ... Treatment of manifestations: Treatment of severe metabolic acidosis with bicarbonate therapy; hyperammonemia (which may be related to severe acidosis or low ATP from impaired aerobic oxidation) may be addressed by treatment of the metabolic acidosis and/or consideration of hemodialysis. ... Two affected individuals have had moderate hyperammonemia in the setting of profound neonatal metabolic stress, potentially related to their severe metabolic acidosis and/or low ATP secondary to impaired aerobic oxidation. ... Low PDC activity has also been noted in lymphocytes as well as liver and skeletal muscle [Ferdinandusse et al 2015, Bedoyan et al 2017]. ... It may be secondary to metabolic acidosis, as it has only been observed in children with profound metabolic acidosis or due to low ATP secondary to impaired aerobic oxidation.
-
Vasculitis
Wikipedia
Laboratory Investigation of Vasculitic Syndromes [13] Disease Serologic test Antigen Associated laboratory features Systemic lupus erythematosus ANA including antibodies to dsDNA and ENA [including SM, Ro (SSA), La (SSB), and RNP] Nuclear antigens Leukopenia, thrombocytopenia, Coombs' test, complement activation: low serum concentrations of C3 and C4, positive immunofluorescence using Crithidia luciliae as substrate, antiphospholipid antibodies (i.e. anticardiolipin, lupus anticoagulant, false-positive VDRL) Goodpasture's disease Anti-glomerular basement membrane antibody Epitope on noncollagen domain of type IV collagen Small vessel vasculitis Microscopic polyangiitis Perinuclear antineutrophil cytoplasmic antibody Myeloperoxidase Elevated CRP Granulomatosis with polyangiiitis Cytoplasmic antineutrophil cytoplasmic antibody Proteinase 3 (PR3) Elevated CRP Eosinophilic granulomatosis with polyangiitis perinuclear antineutrophil cytoplasmic antibody in some cases Myeloperoxidase Elevated CRP and eosinophilia IgA vasculitis (Henoch-Schönlein purpura) None Cryoglobulinemia Cryoglobulins, rheumatoid factor, complement components, hepatitis C Medium vessel vasculitis Classical polyarteritis nodosa None Elevated CRP and eosinophilia Kawasaki's Disease None Elevated CRP and ESR In this table: ANA = Antinuclear antibodies, CRP = C-reactive protein, ESR = Erythrocyte Sedimentation Rate, ds DNA = double-stranded DNA, ENA = extractable nuclear antigens, RNP = ribonucleoproteins; VDRL = Venereal Disease Research Laboratory Treatment [ edit ] Treatments are generally directed toward stopping the inflammation and suppressing the immune system.SERPINA1, CBL, MOCOS, IL18, PRTN3, ADA2, IL10, HLA-B, PTPN22, MEFV, MS4A1, TLR4, HLA-DPB1, NLRP3, CTLA4, MYD88, TNFRSF13C, IL12B, FAS, NFKB1, CFI, ARPC1B, PRKCD, RASGRP1, KLRC4, WIPF1, WAS, SCN11A, TNFSF12, SCN9A, NFKB2, TNFRSF1A, HLA-DPA1, TNF, MVK, NFKBIL1, MLX, STAT4, SLC22A4, HLA-DRB1, SCN10A, VPS13A, CD81, TNFRSF13B, IL23R, UBAC2, IL12A, IFIH1, C4A, CIITA, MPO, CR2, CASP10, ERAP1, CD244, ICOS, CD19, IL1B, SAMHD1, IL12A-AS1, LMX1B, CCR1, PADI4, FASLG, IGAN1, SMUG1, IL1A, IL6, IL1RN, IFNG, CCL2, RBM45, ACR, GYPA, ITGB2, PTX3, ALB, KIR2DS2, CD274, CRP, ICAM1, LAMP2, ADA, WG, KRT20, CD28, GCA, IGHA1, VWF, VEGFA, VCAM1, S100A9, FCGR3B, PDCD1, CLEC6A, S100A12, SH2D1A, LOC102723407, CCL5, LAMC2, ABCC11, BTG3, MMP9, CMAS, NOS3, SLC6A2, TLR2, S100A8, HAMP, LOC102724971, CX3CR1, IL2, CXCR3, CFH, EPHB1, ELK3, CCR5, HMGB1, CSF2, FCGR2A, CXCL10, AHSA1, PYCARD, IKZF1, TNIP1, CD22, CD1E, ADIPOR1, CSK, TLR7, IL23A, CD1D, RTEL1, CAV1, CAST, TLR9, CYCS, TREM1, CAPS, IGHV3-52, CD226, PALD1, CP, CSF3, CXCL13, PALLD, NXF1, MAPK14, CRK, SEC14L2, CCL26, LAMP3, IL17RA, RASGRP3, CD79A, RNF19A, FBLIM1, POLDIP2, CD40, CD40LG, ISG20, CALR, SLC15A4, DEFB104A, APOA2, PLB1, ANPEP, STING1, SLC26A5, SERPINA13P, MIRLET7B, MIR145, MIR320A, ANGPT2, DEFB104B, CCR2, AKR1B1, DEFB4B, MICA, RPL17-C18orf32, AHR, AGT, STS, ATD, WDR11, C1QTNF1, ZEB2, C5AR1, MUC13, GOPC, CD177, SEMA6A, C5, IL21, SERPING1, C1QBP, CLEC7A, ADIPOR2, LPAL2, TUBA1C, BPI, TSLP, IL33, BLK, BCL2, IL26, SLC9A3R2, SPATA2, MAPK8, PLAT, PLG, PMP22, PNN, PODXL, POMC, PRKCB, MAPK1, CXCL1, HLA-C, RPL9, RPL17, RPL31, RPS3A, RXRB, GAS6, FN1, FLNB, PLA2G1B, HLA-DQA1, FCGR3A, IFIT3, LEPR, IRF5, LSAMP, MBL2, IL17A, IL5, MMP2, IL2RA, TNC, PAEP, HSP90AA1, HLA-DRB4, NGF, NOS2, HLA-DQB1, NOTCH4, NT5E, P4HB, FCN1, SCP2, ISG15, CYP2C9, ATN1, DEFB4A, ACE, YWHAZ, PXDN, AIMP2, FGF23, CDR3, GEMIN2, EGF, USO1, CYP2C19, P4HA2, SCAF11, CD83, CD163, LEP, GRAP2, LPAR1, TYK2, FCGR2B, ABCC8, CCL3L1, SH3BP2, F8, F5, SMN1, SMN2, SNCA, EXT1, SYK, TPMT, TADA2A, ETS1, TGFB1, TIMP4, ENO1, TLR5, ELANE, TNFRSF1B, SERPINA3

-
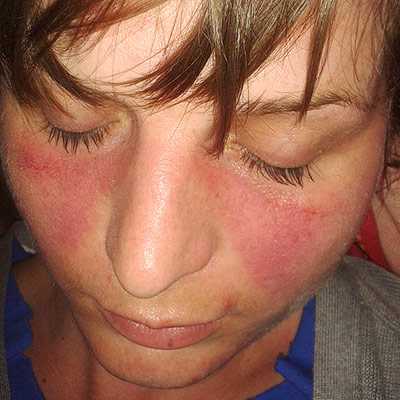
Lupus
Wikipedia
When occurring in conjunction with other signs and symptoms, however, they are considered suggestive. [12] While SLE can occur in both males and females, it is found far more often in women, and the symptoms associated with each sex are different. [6] Females tend to have a greater number of relapses , a low white blood cell count , more arthritis , Raynaud's phenomenon , and psychiatric symptoms . ... SS-A and SS-B confer a specific risk for heart conduction block in neonatal lupus. [74] Other tests routinely performed in suspected SLE are complement system levels (low levels suggest consumption by the immune system), electrolytes and kidney function (disturbed if the kidney is involved), liver enzymes , and complete blood count . ... Malar rash (rash on cheeks); sensitivity = 57%; specificity = 96%. [78] Discoid rash (red, scaly patches on skin that cause scarring); sensitivity = 18%; specificity = 99%. [78] Serositis: Pleurisy (inflammation of the membrane around the lungs) or pericarditis (inflammation of the membrane around the heart); sensitivity = 56%; specificity = 86% (pleural is more sensitive; cardiac is more specific). [78] Oral ulcers (includes oral or nasopharyngeal ulcers); sensitivity = 27%; specificity = 96%. [78] Arthritis : nonerosive arthritis of two or more peripheral joints, with tenderness, swelling, or effusion; sensitivity = 86%; specificity = 37%. [78] Photosensitivity (exposure to ultraviolet light causes rash, or other symptoms of SLE flareups); sensitivity = 43%; specificity = 96%. [78] Blood—hematologic disorder— hemolytic anemia (low red blood cell count), leukopenia (white blood cell count<4000/µl), lymphopenia (<1500/µl), or low platelet count (<100000/µl) in the absence of offending drug; sensitivity = 59%; specificity = 89%. [78] Hypocomplementemia is also seen, due to either consumption of C3 [79] and C4 by immune complex-induced inflammation or to congenitally complement deficiency, which may predispose to SLE. ... These may subside if and when the large initial dosage is reduced, but long-term use of even low doses can cause elevated blood pressure and cataracts . ... The treatment plan for these people requires anticoagulation. Often, low-dose aspirin is prescribed for this purpose, although for cases involving thrombosis anticoagulants such as warfarin are used. [96] Management of pregnancy Further information: Systemic lupus erythematosus and pregnancy While most infants born to mothers who have SLE are healthy, pregnant mothers with SLE should remain under medical care until delivery.DNASE1, FCGR2B, PTPN22, PDCD1, TREX1, IFIH1, TNIP1, IL10, HLA-DRB1, BANK1, ETS1, IRF5, ITGAM, BLK, STAT4, C4A, CTLA4, TNFAIP3, CR2, C1QA, PXK, JAZF1, RASGRP1, TNFSF4, C4B, PRDM1, DEF6, HLA-DQA1, SPP1, FCGR2A, IRAK1, MECP2, UBE2L3, PHRF1, DNASE1L3, C2, SLC15A4, UHRF1BP1, CLEC16A, C1R, RASGRP3, KIAA0319L, FAS, C1S, IL21, CFB, TLR5, FCGR3B, IL4, CAT, CRP, PRL, IL6, ATG5, IKZF1, NCF1, CLU, CD226, IL12B, IL21R, IRAK4, IKZF3, DNASE2, CSK, C1QB, LYN, PTGS2, SIGLEC6, TCF7, P2RY12, ANXA3, RO60, PCNX3, SH2B3, SYNGR1, TLR7, CD40LG, TERT, TNFRSF13B, CYBB, PTPRC, JAK1, SNRPD1, CDKN1A, TIMD4, JUNB, PPARG, LBR, GADD45A, BECN1, TRAF3IP2, RUBCN, ATG7, MAN2A1, INPP5D, RC3H1, PLD4, EP300, LTA, MIR196A2, POLB, IL2RA, CD40, RXRA, HLA-A, IRF7, MTA2, TYK2, CD247, FASLG, C3, RASSF5, IL23R, NCF2, HLA-B, ITGAX, BACH2, CD80, WDFY4, ELF1, TMEM39A, PRKCD, SYT1, GTF2IRD1, MSH5, MICB, ARID5B, LBH, TNXB, TNPO3, HLA-DMB, IL19, LRRK2, HLA-DQB2, JAK2, CXorf21, APOM, SKIV2L, CYP21A2, ITPR3, GLT1D1, AFF1, SERPING1, XKR6, TNFSF15, GRB2, PRRC2A, ENG, PTPN11, NFKBIL1, NMNAT2, NOTCH4, PNP, DRAM1, MPIG6B, DAG1, SMG7, MFHAS1, SLC5A11, HLA-DRA, HLA-DQA2, ERBB2, SMAD3, PEPD, BTNL2, LRRC18, COG6, ATXN2, CASP10, IL12RB2, ATG16L1, HCP5, GALC, FAM98B, MIF, LPP, HLA-DPB1, FCRL3, TNFRSF13C, GABBR1, HIP1, MBL2, MGAT5, LINC01845, GPR19, C1QTNF6, TPI1P2, AHNAK2, FAM86B3P, GPR35, LEP, LURAP1L, NR3C1, GEM, HLA-G, LCP1, IL1B, IFNA1, MUCL3, IFNA13, IFNG, PPP1R18, PSORS1C1, IL1A, IL1RN, HLA-DQB1, IL2, ITGAL, CXCL8, ISG20, IL17A, IL18, CXCL10, IFN1@, IRF8, SCAMP5, KIR3DL1, LAMC2, HLA-DRB9, ZFP90, HLA-L, HMGB1, SPPL3, RTKN2, ARMC3, PUSL1, RBM45, SAMD9L, SPRED2, ICA1, ICAM3, KIT, MMP9, PRKG1, ANKRD44, ANKRD30A, H2BC15, CDR3, KMT2D, NIN, NELFE, IGF2-AS, GPANK1, BAG6, ST8SIA4, EVI5, ZSCAN26, ZNF76, TIMMDC1, TLR8, TAOK3, DDX41, IL23A, VEGFA, VDR, CDHR5, TLR9, TPP2, TNFRSF1B, TNF, TLR4, KRT20, AHI1, FOXP3, MBL3P, IL22, CEPT1, TRIM31, ATXN2L, BTG3, EHMT2, CTRC, IKZF2, MASP2, TNFSF13B, TSBP1, ANP32B, KDM4B, FNBP1, HCG9, DDO, ADGRL2, ANKS1A, CRB1, KALRN, SKAP2, STK19, IL18RAP, CCDC113, ADAM15, TNFSF13, ZNRD1, BARX2, TGFB1, TRBV20OR9-2, TCP11, RNF39, ZSCAN31, SMYD3, ILRUN, PAPOLG, PLD2, PLCL1, PLAT, ABHD8, CENPU, ANKRD55, PBX2, SCUBE1, SLC44A4, ERAP2, VWA7, LY6G6C, NOTCH2, NOS3, OR5V1, OR12D3, FAM167A-AS1, ATRIP, MIEN1, PGBD1, NKD1, AP5B1, CARD9, MTOR, TCF19, CARMIL1, SUOX, STAT3, STAT1, SSB, TRIM21, SRI, SPINK1, NAT2, NADSYN1, SMPD1, SLC12A1, RNPC3, CCL2, MAPK1, ATXN1, RPS20, CAMK1D, ZMIZ1, RNF5, PRR12, RAD51B, RAB4A, PVT1, PRSS1, TRAPPC11, GPSM3, FUT2, BTN2A1, MS4A1, INS-IGF2, ARHGAP4, MIR155, IFNG-AS1, CD70, ALB, DGKQ, DARS1, ACE, HCG18, SFTA2, FCGR3A, ESR1, CD19, DECR1, CDKN1B, LINC01250, MUC21, HCG17, IL12A-AS1, SCARB1, LINC00993, TSBP1-AS1, F3, APOH, TRIM26BP, LINC00243, CD28, IRF1-AS1, LINC01185, C1orf141, OR14J1, MIR146A, EPHB2, ATP6V1G2, STX17-AS1, MSH5-SAPCD1, EHMT2-AS1, CREM, LINC01088, MIR210HG, ACR, MIR21, SBK1, ZKSCAN4, ADCY7, LYST, HNRNPA1P1, RNU4-36P, CFTR, LINC01511, DNMT1, ABCF1, AP4B1-AS1, WAKMAR2, CALR, BTK, CR1, BCL2, CSNK2B, C8A, CREBL2, KLK3, CD44, F5, CCL5, PON1, ABCB1, CXCL13, GSTT1, CD27, SLAMF1, IL9, CAMK4, DNMT3A, IFNB1, CXCR5, CD22, TP53BP1, BCR, HPGDS, CD14, FLI1, ITGB2, HLA-C, EZH2, FAM167A, CD38, SH2B2, SERPINE1, F2, MTHFR, IL37, CFH, CD274, ICAM1, ICOS, AQP4, PTPA, CXCL12, SH2D1A, CD69, MAVS, MPO, LSM2, APOL1, CXCR4, CXCR3, SOAT1, KIR3DL2, HRES1, AIM2, NLRP3, TPMT, MFGE8, TRAF1, TLR3, TLR2, TAP2, P2RX7, HSPA4, ADIPOQ, GSTM1, VCAM1, PRKCB, TP53, GRIN2A, FCN2, IGHG3, HT, SNCA, TPO, TNFRSF1A, SNRNP70, CCR5, AGT, NFKB1, CD24, KIR2DS1, PPP2CA, IFI44L, CD244, TNFRSF17, MIR125A, REN, WG, HAVCR1, MBD2, MEFV, HSPA1A, APOE, MYDGF, HSP90AA1, SLAMF6, ANXA1, SOCS1, IFI16, PADI4, TBX21, LY9, PTX3, IFNL1, IL33, CYP2D6, PECAM1, PSMA5, CD46, OCA2, RPL17, JUN, LY6E, CD86, IL17F, SRSF1, FLII, CYP2B6, CYP1A1, S100B, CCR7, MBP, H4C15, ENAH, CSF2, SOD1, SELE, H4-16, TAP1, CD72, HAVCR2, PIK3CD, CST3, RFX1, SELL, LCN2, IGAN1, TG, LGALS3, IRF1, ARHGEF5, MICA, GPI, AHR, B2M, HDAC6, TAM, H4C9, FCGR1A, H4C1, H4C4, H4C6, H4C12, H4C11, H4C3, H4C8, SIRT1, H4C2, H4C5, H4C13, H4C14, MAP4K3, GTF2I, GZMB, AP5Z1, SPATA2, HSPA1B, ANXA6, FCGR2C, RPL17-C18orf32, TIMELESS, LILRB1, IL15, ADA, FOS, IL16, BDNF, XRCC1, GATA3, TNFRSF4, ACTB, PCNA, HDAC9, AMH, MIR326, H3P28, UBASH3B, LOC102724971, ARMH1, MNAT1, LOC102723407, HLA-DMA, EBNA1BP2, SAMHD1, TNFRSF11B, ARHGEF2, S100A9, PBX1, IL27, ISG15, FOXO1, GSTP1, ADAR, NOD2, RABEPK, LANCL1, GAS5, AKT1, GAS6, GRN, MBD4, PSMD7, SERPINA1, PLG, PIK3CG, KLRK1, PIK3CB, PIK3CA, SLAMF7, NLRP1, CBLIF, ATN1, CIITA, KLRC4-KLRK1, SDC1, IFIT1, MIR150, TP63, CD180, ZAP70, IFNAR1, IL12A, SYK, IFNAR2, SELP, LGALS9, SIGLEC1, RPSA, IL10RA, KIR2DS2, VWF, CASP8, EGR2, C1QC, MDM2, CAMP, MIR142, TNFRSF6B, IRF3, TREM1, IL17D, PARP1, HSP90B1, SEMA3A, GORASP1, STAT5A, PRF1, ROBO3, SERPINB2, PSIP1, STAT5B, GDE1, RUNX1, LILRA3, CASP1, TRAF6, UBASH3A, TGFB3, WNK1, PSS, CCR6, GGCT, CASP3, C4B_2, TNFRSF8, UPK3B, CDR1, TNFSF10, DDX58, AICDA, IL18R1, IGHV3-52, CDK5R1, PELI1, PDLIM7, APCS, S100A8, CCL3L1, MTMR3, SOCS3, APOA1, TPI1, CD48, RELA, REL, F11R, CDKN2A, SNRPA, EBI3, BCHE, SERPINB3, SLC22A4, SLC6A2, TNFSF12, SIGIRR, HNRNPDL, HAMP, TNFSF11, PLAAT4, OASL, COX8A, CXCL16, KLRC1, CFHR5, HLA-DRB5, EMB, IFNA2, HLA-DOA, SLCO6A1, GCK, FOSB, KIR2DL5B, MX1, HIF1A, LAIR1, DNMT3B, KLRC2, HGF, MIR34A, NFKBIA, STING1, IL6R, IFNA17, IFNGR1, MAP1LC3B, IL4R, ITPA, ARID3A, IFI6, XRCC6, MRC1, CLEC4C, JUND, IFIT3, ELOF1, CTNNB1, EPHB1, HNRNPC, CGAS, PDCD1LG2, EFNA2, HP, CYP21A1P, IL13, LGALS3BP, ELK3, FCER1G, LGALS1, LEPR, EGFR, CREB1, OAZ1, CSF1, EGR3, BTLA, GCHFR, CDCA5, ESR2, GSTK1, LDLR, IFNL3, HSPA2, RMDN3, CD1D, ZPBP2, ERN1, KLRD1, IER3, KRAS, LBX1-AS1, BIN1, SMUG1, STAT2, ROCK2, SHOC2, IFNK, CFHR1, MIR31, ATF7IP, NR1I3, H1-0, SYBU, MIR17HG, ABCD1, GIMAP5, ERVK-20, SLC6A4, CD84, SLC11A1, SLC19A1, BMS1, FBXW7, RMDN2, RMDN1, CHAF1B, IKBKE, NR1I2, SNRPB, SNRPC, SEC14L2, LAG3, SOD2, CD4, SP1, SPG7, CCR2, KIR2DL2, CASR, SLEB4, E2F1, TRAF2, MIR22, TRPC6, TRPM2, TSPO, PWAR1, MIR483, EIF4E, XPR1, HRAS, MANF, AIRE, IL34, IL7R, ELANE, MIR126, EDN1, SGSM3, KLF13, MIR200A, CXCR2, VIP, VPREB1, IL10RB, MIR20A, WIPF1, TNFRSF9, XRCC3, HMOX1, HLA-DRB4, MIR223, C5, HLA-DRB3, NEAT1, ADAM17, SS18L1, MIR29B2, CRBN, ZEB1, KIR2DL3, CAST, CNOT8, NR1H4, KIR2DL1, TGFB2, KDR, CD163, LAP, THBS1, CALM3, TIMP1, MIR29B1, EIF4EBP1, CALM2, CALM1, F2RL1, F2R, CYCS, BCL6, ITGB1, TSC22D3, IL32, LGALS8, MIR19B1, TNFSF12-TNFSF13, IL20, NFATC2, ARR3, ADAM10, PRKAR1A, PLA2R1, PRKCA, CECR, CEBPB, CNOT7, IFNGR2, CD52, NCAM1, SLC39A8, VSIR, EIF2AK2, SEMA7A, ERVK-6, GABPA, IFI44, MYD88, DKK1, MASP1, MTR, LPA, SRCIN1, MIR148A, LOC107987479, NR0B2, PTEN, NFE2L2, PPARD, TNFRSF25, IL24, TNFSF9, CREBBP, DEFA1A3, PF4, PGF, AR, IFI27, FN1, SLC52A2, ACACA, ENO1, ERVW-1, NOTCH1, NOS2, CFHR3, PLA2G1B, NM, CCR4, CCR1, NHS, CLTA, IL25, CXADRP1, HSP90AB1, NGF, TNFRSF14, PON3, ACP5, POU4F1, MIR663A, TRIM38, LILRB4, RPS19, TRAP1, MAP4K1, SLC12A9, ABCA1, MDGA2, HTR1A, ICOSLG, CD276, PRRT2, S100A10, RIPK1, S100A12, CORO1A, CXCL9, CHAF1A, CD33, CYP2C19, CCL3, P2RY2, CYP2E1, P2RY1, MDK, RETN, VGLL3, DBA2, CYP3A5, CX3CL1, TIRAP, MIR181A2, G3BP1, MMP2, NT5C1A, IFNLR1, MERTK, PTPN2, PTPN6, ADM, RIPK3, ZNF423, CD74, MT1G, AGER, POTEF, KDM6B, LILRB2, RAG2, ATG16L2, SLEH1, TBC1D9, IGF1, MIR302D, MPL, MIR15B, RFC1, HFM1, CXADR, KIR2DL5A, RNASE2, TRIM13, ENTPD1, AGTR1, GNAO1, EPO, CENPV, PYCARD, IGHV3OR16-7, IGHV4-34, SETD2, TIGIT, LAMTOR2, IGHV3-69-1, MIR145, CCDC22, PRSS55, IGHV3-7, DLL1, IGKV3-20, TRIM39-RPP21, ULBP2, CABIN1, RBMX, FCRL6, LINC01193, LOC110806262, IL31, ATF6, H3P44, P2RX2, IRGM, AGBL3, PCSK9, TSPAN33, CIC, SUMO4, MPRIP, PHLDB1, NBEAL2, H3C15, MALAT1, LINC02605, PUF60, LCE3B, FSTL1, USP17L2, TREX2, ENHO, POLG2, PADI2, PTGDR2, H3P47, TICAM2, LCE3C, H3P23, PDAP1, TUSC2, SYNPO, H3P8, PERCC1, CCL4L1, TMED10P1, RNF19A, CNTNAP2, POLDIP2, THRIL, ARL5A, PHGDH, ERAL1, EHF, FGF22, LAT, DKK3, IL17B, PDCD4, CNTN6, ADAMDEC1, SGMS1, SLEN3, PRDX5, COTL1, SPDEF, TREML4, KCNH4, USP17L9P, APOA1-AS, BRD4, SEC61G, RSPO1, KLK9, MTREX, MLKL, ZSCAN1, LAMA1, LOC102723971, MILR1, FLRT2, CRYGEP, RIPK4, TBK1, CTHRC1, SCYL1, KIDINS220, CREB3L1, SIGLEC12, USP17L26, PDP2, MRTFA, NBDY, BAGE3, TUBA1C, TRIM63, SPZ1, USP17L25, USP17L24, IL1F10, DEFA1B, NLRC4, HIVEP3, RFH1, ABHD6, RNF185, SLC7A9, SPHK2, SIRPG, CHST12, NLRP2, WDR11, RNF114, EIF2AK4, ALG1, CCL28, BDH2, DNER, USP17L28, BMS1P20, DUSP22, USP17L27, ACKR3, ORMDL3, SLURP1, MUC16, HRH4, MIR654, RPAIN, ZNF419, COL18A1, RNF128, MUL1, TNFAIP8L2, DHX40, VTCN1, UCA1, POU5F1P4, MT1IP, AHNAK, FBXO31, CAMKMT, SLED1, SCD5, NAA25, TSGA10, ZC3H12A, POU5F1P3, RSPH6A, MIR499A, MIR448, CLEC7A, ANO3, MIR633, SH3BGRL3, MIR629, MIR621, DOCK8, ARHGEF28, HNP1, EBF2, MIR525, MIR551B, SLEB3, SNORA12, MIR451A, H3C13, UNC93B1, VKORC1, MIR410, IMPACT, IL22RA2, HILPDA, LARP6, PLB1, MZB1, NT5C3A, C1RL, LINC00513, CASP12, TNFRSF12A, WNT16, NCKIPSD, LINC01672, TMC8, IGLL5, RTRAF, SF3B6, ACSL5, MIR203A, ZNF688, MIR3148, LRP1B, MIR198, EGFL7, MIR196A1, CBLL2, SLEN2, SLEN1, DUOX2, MIR152, MIR5003, MIR5100, ERVW-4, ASAP1, MIR17, MIR183, CLEC4A, IBD5, EXOSC3, ERVK-15, TMED7, GAL, P2RX5-TAX1BP3, ZSWIM2, GPR84, MIR3074, DUOX1, MIR34B, MIR939, MIR873, TRIM68, RBM23, HOTAIR, MTPAP, SIGLEC14, ARL8B, RAVER2, SLC52A1, MIR34C, MIR93, USP17L30, USP17L29, NUDT15, MIR98, DDX19A, LRG1, C20orf181, EARS2, MIR210, TERF2IP, MIR221, OR2AG1, MIR224, NEK7, TMED7-TICAM2, MIR1279, MIR23B, MIR27A, DDIT4, MIR30A, XAF1, DOCK11, H3C9P, DEFB4B, KCNH8, DUSP23, TTC34, H3C14, NAT1, SPI1, RPP14, IGKV@, AGFG1, HPRT1, HNRNPD, HMGN2, HMGN1, HLX, HLA-H, HLA-DPB2, HLA-DPA1, HELLS, HDAC1, HCRT, HCLS1, HCFC1, H2BC5, HES1, HSD11B1, HSPA1L, IFNA5, CCN1, IGFBP3, IGFALS, IGF1R, IGBP1, IFNR, CFI, HSPA5, ICAM4, HSPG2, HSPE1, HSPD1, HSPB2, HSPB1, H2AX, MSH6, GSTM2, FCGRT, FOXO3, FOXJ1, FOXC1, FH, FGF2, FEN1, FCER1A, FLT1, FCAR, FAP, FANCB, PTK2B, ACSL4, F12, FLNB, FLT3, GSN, GCG, GRIN2B, GPX4, GPT, GPER1, GLO1, GFI1, GC, FLT3LG, GBP2, GAPDH, GABRP, FYN, FPR1, FMR1, JCHAIN, IGLV@, F8, IL1R1, MOG, MMP12, MMP11, MMP7, MMP3, MMP1, FOXO4, KMT2A, MFAP1, MEF2D, MEF2A, MCL1, MBNL1, MAGEB2, SMAD7, MPP1, MRE11, ABCC1, MT1L, MTRR, MTNR1A, COX1, ATP6, MT2A, MT1X, MT1M, MSN, MT1JP, MT1H, MT1F, MT1E, MT1B, MT1A, LYZ, LUM, LTK, IRF2, KCNA3, ITGB3, ITGAE, ITGA2, IRF6, IRF4, IRAK2, KIR2DL4, INSRR, IDO1, IL12RB1, IL7, IL5, IL1RAP, KIF5A, KIR2DS3, LMNA, LAIR2, LIG4, LCT, LCP2, LCK, STMN1, LAMC1, AFF3, KIR2DS5, L1CAM, KRT8, KRT1, KNG1, KLRB1, KLK1, F10, EVPL, HNRNPUL1, CETP, KRIT1, CBLB, CBL, RUNX1T1, RUNX2, CASP9, CASP6, CAPG, CALCR, C6, C5AR1, BTF3P11, KLF5, BST2, BSG, CCND3, CCT, CCT6A, CD63, CDKN1C, CDK6, CDH13, CDK1, CDA, CD79B, CD58, CD2, CD36, CD34, CD8B, CD8A, CD5L, CD3E, DST, BMPR2, BID, ADD2, AMPD2, AMELX, ALOX5AP, AHCY, AGTR2, ADRB3, ADARB1, ANXA5, ACTN4, ACP3, ACP1, ACHE, ABL1, ABCA2, ANXA2, APOA4, BGN, ATF3, BGLAP, BCYRN1, BCKDHB, BAX, AXL, ATM, SERPINC1, APOB, ASS1, ARSF, ARSA, ARHGDIB, ABCC6, APRT, CDKN2C, CTSC, ETS2, CGB3, S1PR1, TYMP, EBF1, E2F2, DUT, DUSP4, DTX1, DPP4, DXO, DNTT, DNM1, DMBT1, DLAT, DHFR, DEFB4A, EDNRA, EEF1A2, EFNA5, EMP1, ERV3-1, ERG, ERCC2, EPHX2, EPHX1, EPHA3, MARK2, EIF4EBP2, ELK1, ELF4, ELAVL1, ELAVL2, SERPINB1, EIF4G1, DEFB1, DEFA3, DEFA1, CNR2, CRK, CRH, ATF6B, CP, COMT, COL11A2, CNN3, CRYGD, ABCC2, CMKLR1, CLIC2, CISH, CHRM3, AKR1C4, CRYGC, MAPK14, DHX9, CX3CR1, DDX6, DDT, CYP24A1, CYP19A1, ADAM3A, CYBA, CTSS, CSF3, CTSK, CTSE, CTSB, CTRL, CTBP2, CSF3R, MTX1, GADD45B, MYO9B, SEMA5A, EIF3C, AOC3, HSD17B6, USO1, SCARF1, FCN3, API5, PIK3R3, DHX16, RECK, TAGLN2, PLA2G6, H2BC17, H2BC4, H2BC10, DGAT1, ABCC3, MBTPS1, PROM1, H2BC11, SNURF, EIF2B5, SQSTM1, ASAP2, KAT2B, SOCS2, TNFSF14, HESX1, TNFRSF10B, TNFRSF10C, TNFRSF18, SIGLEC5, RIPK2, H2BC6, H2BC7, H2BC8, UCP2, EZR, VHL, UVRAG, USF1, UMOD, SCGB1A1, UCHL1, VTN, TYRP1, TYROBP, TYR, TYMS, TXNRD1, CRISP2, VIM, XIST, EOMES, USP7, TMEM187, SMC1A, PDHX, AIMP2, LST1, DDX39B, IL1R2, XRCC4, LAPTM5, MZF1, YWHAZ, YWHAB, YY1, XRCC5, HSPB3, PRC1, NBN, SLC7A7, AHSA1, PROCR, RNASEH2A, ZNRD2, NXF1, IRF9, TRIM22, MAEA, STUB1, CDK2AP2, TRIB1, KLRG1, DDX39A, PSME3, CEBPZ, CELF2, DCTN6, B3GNT2, YWHAQ, ADAMTS13, PIM2, LILRA2, LILRA1, SDS, TMED10, SUB1, CCR9, PAPOLA, JTB, MRPS30, C1QL1, CFHR4, CD3EAP, PTPRU, NR1H3, BCL2L11, AIMP1, GRAP2, LONP1, KLF4, CD83, MED14, STK17A, MSC, NCR1, PTTG1, FCMR, DEDD, OSMR, ATG12, P2RX6, NCR2, GSTO1, SCO2, SETD1A, RABGAP1L, DDX46, FCHSD2, ZEB2, RAPGEF5, RASSF2, DCAF1, EIF2AK3, KLK4, IER2, CCL4L2, MACROH2A1, RAB3D, TBPL1, TOP1, ICAM5, TIMP3, THY1, POU5F1, POU4F2, POU2AF1, PON2, PML, PLXNA2, PLSCR1, PLA2G2A, PIN1, PIM1, PIK3C2A, PIGF, SLC25A3, PGM1, PGK1, MED1, PPBP, PPIA, PRLR, PSMB6, PRTN3, PRPS2, PROS1, PRODH, PROC, MAP2K1, PRKAA1, MAPK3, PRKDC, PRKCQ, PRKAR2B, PRKAB1, PRKAA2, PFDN5, PDGFRA, PDE7A, NOS1, OAS1, NUP98, NTRK1, NT5E, NPPA, NPY, NFIL3, OGG1, NF1, NEU1, NEDD9, SEPTIN2, NDUFS4, NCL, OAS2, CLDN11, PDE3B, PAK3, PCSK1, PC, PAX5, PRKN, PAPPA, REG3A, PAH, OXA1L, FURIN, P4HB, P2RX5, P2RX4, P2RX3, P2RX1, PSMB9, PSMD4, PSMD9, SLC2A1, SMN1, SLC22A2, SLC8A1, SLC6A8, SLC4A1, SLC2A4, SLC1A7, SMS, SLC1A5, SKP2, SHBG, SH3BP2, SGK1, SFTPD, SMN2, FSCN1, SET, SUV39H1, THM, THBD, THAS, TGM2, TMBIM6, TAGLN, STAT6, SNRPN, SSRP1, SRP54, SRP19, SPTAN1, SPN, SOS1, SRSF5, SDC2, PSMD12, RAD52, RELB, REG1A, RASGRF1, RASA1, RAG1, RAF1, MOK, RET, NECTIN2, PVR, PTN, PTK2, PTGS1, PTGDS, RENBP, TRIM27, XCL1, SCO1, CCL21, CCL20, CCL19, CCL17, CCL14, CCL4, S100A1, RLN2, RSU1, RPS27A, RPS6, RPLP0, SNORA73A, ABCE1, H3P10
-
Blood Group, P1pk System
OMIM
All NOR+ individuals had at least one P1 allele (42C; 607922.0007) The Q211E mutation broadened the acceptor specificity of the enzyme, causing the transferase to acquire the ability to catalyze the synthesis of Gal(alpha)1-4GalNAc present in NOR-related glycolipids, without losing its ability to transfer the Gal residue to the C4 of Gal (Gal(alpha)1-4). In NOR+ erythrocytes, the mutant enzyme transfers alpha-Gal to the GalNAc of Cb4Cer, transforming a small portion of Gb4Cer into the NOR1 glycolipid.
-
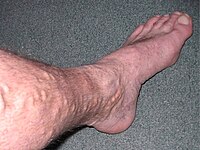
Chronic Venous Insufficiency
Wikipedia
CEAP classification is based on clinical, causal, anatomical, and pathophysiological factors. [9] According to Widmer classification diagnosis of chronic venous insufficiency is clearly differentiated from varicose veins. [10] It has been developed to guide decision-making in chronic venous insufficiency evaluation and treatment. [5] The CEAP classification for CVI is as follows: C0: no obvious feature of venous disease C1: the presence of reticular or spider veins C2: Obvious varicose veins C3: Presence of edema but no skin changes C4: skin discoloration, pigmentation C5: Ulcer that has healed C6: Acute ulcer Etiology Primary Secondary (trauma, birth control pill) Congenital (Klipper trenaunay) No cause is known Anatomic Superficial Deep Perforator No obvious anatomic location Pathophysiology Obstruction, thrombosis Reflux Obstruction and reflux No venous pathology Management [ edit ] Conservative [ edit ] Conservative treatment of CVI in the leg involves symptomatic treatment and efforts to prevent the condition from getting worse instead of effecting a cure.EFEMP1, KCNH8, SKAP2, TGFB1, ICAM1, ITGAL, ITGB2, VCAM1, COL1A2, SGSM3, CLDN1, VEGFA, CLDN5, TIMP2, PIK3CD, MAPK3, PIK3CG, ENG, PIK3CB, PIK3CA, MMP12, MMP9, MMP2, MMP1, BLOC1S2

-
Nephritic Syndrome
Wikipedia
By contrast, nephrotic syndrome is characterized by proteinuria and a constellation of other symptoms that specifically do not include hematuria. [6] Nephritic syndrome, like nephrotic syndrome, may involve low level of albumin in the blood due to the protein albumin moving from the blood to the urine. [7] Contents 1 Signs and symptoms 2 Causes 2.1 Children/adolescents 2.2 Adults 3 Pathophysiology 4 Diagnosis 4.1 Physical examination 4.2 Laboratory testing 4.3 Invasive testing 5 Treatment 6 Prognosis 7 Epidemiology 7.1 Geography 7.2 Gender 7.3 Race and ethnicity 7.4 Other countries of world 8 References 9 Further reading 10 External links Signs and symptoms [ edit ] Historically, nephritic syndrome has been characterized by blood in the urine ( hematuria ), high blood pressure ( hypertension ), decreased urine output <400 ml/day ( oliguria ), red blood cell casts , pyuria , and mild to moderate proteinuria . [8] [9] If the condition is allowed to progress without treatment, it can eventually lead to azotemia and uremic symptoms. [9] This constellation of symptoms contrasts with the classical presentation of nephrotic syndrome (excessive proteinuria >3.5 g/day, low plasma albumin levels ( hypoalbuminemia ) <3 g/L, generalized edema , and hyperlipidemia ). [8] [10] Signs and symptoms that are consistent with nephritic syndrome include: Hematuria ( red blood cells in the urine ) [11] Proteinuria (protein in the urine) ranging from sub- nephrotic (<3.5 g/day) to >10 g/day, [7] although it is rarely above nephrotic range proteinuria levels. [12] Hypertension [13] resting blood pressure is persistently at or above 130/80 or 140/90 mmHg. [14] Blurred vision [4] Azotemia (increased plasma Urea and Creatinine ) [2] Oliguria (low urine output <400 ml/day) [2] Red blood cell casts (seen with urine analysis and microscopy ) [15] Pyuria ( white blood cells or pus in the urine) [15] Causes [ edit ] Purpura Nephritic syndrome is caused by extensive inflammatory damage to the glomerulus capillaries , which is associated with a variety of medical conditions that we will discuss. ... If positive, then the physician may order additional tests to determine which autoimmune condition is the cause and how best to treat it. [35] Antiglomerular basement membrane (anti-GBM) antibody - If positive, this is highly indicative of Goodpasture's syndrome and can be used to guide treatment. [9] Antineutrophil cytoplasmic antibody (ANCA) - If positive, this indicates that there is likely an underlying vasculitis that may be causing the acute nephritic syndrome. [36] Serum complement (C3 and C4) - Complement factors bind to antibodies to form immune complexes and a decreased serum complement level could indicate that the complement is being consumed at a higher rate due to the formation of immune complexes leading to deposition in the glomerulus of the kidney. [9] Invasive testing [ edit ] A kidney biopsy will provide a fully definitive diagnosis of nephritic syndrome and may also reveal the underlying cause of the nephritic syndrome depending on the underlying pathological process. ... Some treatment modalities commonly used to meet these goals include: Bed rest during the recovery process to ensure administration of optimal medical therapy with as low of a risk as possible for any exacerbating factors (falls, infection , etc). [38] Fluid restriction to minimize the risk of edema (if not already present) or to reduce any active edema that may be present. [39] A special diet during the hospital stay that restricts sodium , potassium , and fluids in conjunction with the previously mentioned fluid restriction in an attempt to control symptoms of fluid overload . [40] Administration of diuretics if patient is showing signs of fluid overload .
-
Alcohol-Related Dementia
Wikipedia
The presence of the Apolipoprotein c4 allele. [12] Treatment [ edit ] If the symptoms of alcohol dementia are caught early enough, the effects may be reversed.
-
Systemic Inflammatory Response Syndrome
Wikipedia
Critical Care Medicine . 35 (9 Suppl): S584–90. doi : 10.1097/01.CCM.0000279189.81529.C4 . PMID 17713413 . ^ Rinaldi, S; Landucci, F; De Gaudio, AR (September 2009).TNFRSF1B, NOS2, IL6, TNF, CRP, IL10, IL1B, ALB, BTBD8, TLR4, CXCL8, RIPK1, MBL2, HMGB1, TLR2, HGF, HMOX1, LCN2, FCGR1A, IFNG, MASP2, RIPK3, CYBB, CD14, CCL2, THBD, SERPINE1, SCN10A, DCAF1, SELE, SELL, ZFYVE9, SELPLG, SRSF5, SPATA2, PPP6R2, CIR1, SLPI, SPG7, TNFAIP3, HGS, SPP1, ARTN, NRP1, SST, TCF21, SERPINB7, FCN3, TAM, LAT2, VIP, EDIL3, UROD, TNNI3, NR1I3, ADM, TRIM13, STIM2, COLEC11, CORO7, ASRGL1, ZBP1, RPAIN, CHRFAM7A, IL33, SPECC1, NLRP3, SLC18B1, STON1-GTF2A1L, ANKRD42, MIR143, MIR15A, MIR191, MIR23B, CXADRP1, TLX1NB, LOC100505909, NOD2, RTN4, CXCL13, NAT10, SH2B2, CCL27, RSC1A1, GTF2A1L, STON1, CARD8, SIRT2, ARC, PADI4, CLEC5A, LY96, ATRNL1, C5AR2, PDLIM3, CD274, IL19, IRAK4, GDE1, TLR9, RTN1, PIP, RRBP1, CPN1, CXADR, ATN1, EDA, EDN1, ESR1, F2, F3, FKBP1A, FKBP1AP1, FKBP1AP2, FKBP1AP3, FKBP1AP4, FPR1, GABPA, NR3C1, GRP, GSN, CSF2, CISH, CFH, CHI3L1, AFM, AGRP, ALDH2, ALPP, BIRC2, KLK3, ARR3, ATHS, BPI, C5AR1, CALCA, CAPN1, CARS1, CASP1, CASR, CD28, CDK9, HARS1, HP, PTX3, MIF, NFE2L2, NFKB1, NPPB, OTC, PF4, SERPINA1, PIK3CA, PIK3CB, PIK3CD, PIK3CG, ADRB2, PLG, PPBP, PPIA, PRKAR1A, PTEN, PTH, COX1, MEFV, AGFG1, MC1R, HRG, HSPA1A, HSPA1B, HSPA4, HSP90AA1, DNAJC4, ICAM1, IL1A, IL1RN, IL4, CXCR1, ITGB1, ITGB2, KCNJ5, LAMC2, LEP, LTA, MIR4772
-
Muscular Dystrophy-Dystroglycanopathy (Congenital With Brain And Eye Anomalies), Type A, 4
OMIM
Mutation in the FKTN gene can also cause a less severe congenital muscular dystrophy-dystroglycanopathy without mental retardation (type B4; MDDGB4; 613152) and a limb-girdle muscular dystrophy-dystroglycanopathy (type C4; MDDGC4; 611588). Description MDDGA4 is a severe autosomal recessive muscular dystrophy-dystroglycanopathy with characteristic brain and eye malformations, seizures, and mental retardation. ... Although clinical details were limited, the patient had infantile onset, muscle hypertrophy, increased serum creatine kinase, and low IQ. He only achieved sitting. There were no eye abnormalities, but brain MRI showed cerebellar cysts, white matter abnormalities, and hydrocephalus. ... Cotarelo et al. (2008) described a Spanish female infant, born of nonconsanguineous parents, who was diagnosed with Walker-Warburg syndrome and died on day 5 of life after suffering respiratory apnea and bradycardia. She had a dysmorphic face with low-set malformed ears, left preauricular tag, thoracic hemivertebrae, and cardiac defects. ... Matsumura et al. (1993) reported that dystrophin-associated proteins such as alpha-dystroglycan (DAG1; 128239) have abnormally low expression in FCMD. DAG1 is a cell surface protein that plays an important role in the assembly of the extracellular matrix in muscle, brain, and peripheral nerves by linking the basal lamina to cytoskeletal proteins. ... INHERITANCE - Autosomal recessive HEAD & NECK Eyes - Optic atrophy - Retinal detachment - Abnormal eye movements - Strabismus - Myopia - Hyperopia - Cataracts - Microphthalmia - Retinal dysplasia (1 patient) CARDIOVASCULAR Heart - Myocardial fibrosis - Dilated cardiomyopathy (onset in second decade) - Cardiac defects (reported in 1 patient) - Atrial septal defect - Double subaortic ventricular defect - Hypoplastic left ventricular outlet - Pulmonary stenosis - Transposition of the great arteries RESPIRATORY - Respiratory insufficiency SKELETAL - Contractures, progressive Spine - Spinal rigidity - Scoliosis MUSCLE, SOFT TISSUES - Muscular dystrophy - Hypotonia - Muscle atrophy - Calf muscle hypertrophy - Muscle biopsy shows decreased glycosylation of alpha-dystroglycan (DAG1, 128239 ) NEUROLOGIC Central Nervous System - Mental retardation - Poor motor development - Polymicrogyria - Leptomeningeal thickening - Focal interhemispheric fusion - Low density white matter on CT scan - Cobblestone lissencephaly - Pachygyria - Polymicrogyria - Agyria - Agenesis of the corpus callosum - Encephalocele (rare) - Hydrocephalus - Cerebellar cysts - White matter changes - Seizures - Hyperekplexia (rare) - Pyramidal tract hypoplasia - Brainstem hypoplasia - Cerebellar hypoplasia - Holoprosencephaly (1 patient) Peripheral Nervous System - Hypo- or areflexia LABORATORY ABNORMALITIES - Increased serum creatine kinase MISCELLANEOUS - Onset in infancy - Incidence of 1 per 10,000 births in Japan MOLECULAR BASIS - Caused by mutation in the fukutin gene (FKTN, 607440.0001 ) ▲ CloseFKRP, POMGNT1, POMT1, DAG1, FKTN, CRPPA, POMGNT2, B4GAT1, POMT2, LARGE1, RXYLT1, COL4A1, POMK, B3GALNT2, GMPPB, B3GNT2, ZC4H2, ALG1, LAMA2, EGF, ANO5

-
Mosquito Bite Allergy
Wikipedia
Mosquito bite allergies are informally classified as 1) the Skeeter syndrome , i.e. severe local skin reactions sometimes associated with low-grade fever; 2) systemic reactions that range from high-grade fever, lymphadenopathy , abdominal pain, and/or diarrhea to, very rarely, life-threatening symptoms of anaphylaxis ; and 3) severe and often systemic reactions occurring in individuals that have an Epstein-Barr virus-associated lymphoproliferative disease , Epstein-Barr virus-negative lymphoid malignancy , [2] or another predisposing condition such as Eosinophilic cellulitis or chronic lymphocytic leukemia . [3] The term papular urticaria [4] is commonly used for a reaction to mosquito bites that is dominated by widely spread hives . ... In subsequent mosquito bites, IgE and IgG appear involved in the development of both immediate and delayed skin reactions while T cells appear involved in development of the delayed skin reactions. [8] The acquired IgE binds mosquito saliva proteins and then triggers skin tissue cells such as mast cells to release at least two mediators of allergic reactions, histamine and leukotriene C4 . These mediators contribute to the development of the wheal, itch, and other components of the immediate reaction. ... DEET or permethrin ) are effective, highly recommended means for reducing mosquito bites. [6] Daily doses of a non-sedating second-generation anti-histamines (e.g. cetirizine or levocetirizine ) can effectively reduce the immediate and delayed reactions to mosquito bites. [8] The use of recombinant mosquito saliva proteins to desensitize individuals against developing reactions to mosquito bites has yielded variable results and requires further study. [6] Treatment [ edit ] Treatment of ordinary small or large mosquito bite reactions is limited to the use of non-sedative H1 antihistamines , e.g. cetirizine [6] or a drug with combined activity in inhibiting histamine and platelet-activating factor , e.g. rupatadine . [9] Randomized, double-blinded, placebo-controlled studies are needed to determine if antileukotriene drugs or topical steroids have beneficial effects in reducing the symptoms of these bites. [6] Skeeter syndrome reactions [ edit ] Main article: Skeeter syndrome Presentation [ edit ] The Skeeter syndrome is by definition a mosquito bite allergy that consists of a large mosquito bite reaction that may be accompanied by a brief or longer-term (i.e. days to weeks) low-grade fever. [8] and, on rare occasions, vomiting. [10] The bite site shows an intense, large reaction often resembling a cellulitis infection that persists for days to weeks. [5] The syndrome usually afflicts healthy children, immune-deficient persons, and individuals who are new to an area inhabited by mosquito species to which they have not been exposed. [6] Pathophysiology [ edit ] Mechanistically, the Skeeter syndrome appears to be a particularly intense variant of the ordinary mosquito bite reaction. ... In addition to second generation, non-sedative H1 antihistamines, antipyretics and nonsteroidal anti-inflammatory drugs are typically used to treat patients with acute attacks of the syndrome. [6] Systemic allergic reactions [ edit ] Presentation [ edit ] Individuals with systemic mosquito bite allergies respond to mosquito bites with intense local skin reactions (e.g. blisters, ulcers, necrosis, scarring) and concurrent or subsequent systemic symptoms (high-grade fever and/or malaise ; less commonly, muscle cramps , bloody diarrhea, bloody urine, proteinuria , and/or wheezing ; [3] or very rarely, symptoms of overt anaphylaxis such as hives , angioedema (i.e. skin swelling in non-mosquito bite areas), shortness of breath, rapid heart rate, and low blood pressure]]. [8] There are very rare reports of death due to anaphylaxis following mosquito bites. [6] Individual with an increased risk of developing severe mosquito bite reactions include those experiencing a particularly large number of mosquito bites, those with no previous exposure to the species of mosquito causing the bites, and those with a not fully developed immune system such as infants and young children. [8] Individuals with certain Epstein-Barr virus-associated lymphoproliferative , [12] non-Epstein-Barr virus malignant lymphoid, [2] or other predisposing disease [3] also have an increased rate of systemic mosquito bite reactions but are considered in a separate category (see below).

- Chst3-Related Skeletal Dysplasia GeneReviews
-
Chondrodysplasia Punctata 2, X-Linked
GeneReviews
When the parents are clinically unaffected, the risk to the sibs of a proband appears to be low but greater than that of the general population. ... Other distinctive features include downslanting palpebral fissures, hypertelorism, low-set ears, and high-arched palate [Happle 1979, Herman 2000]. ... RCDP2 (OMIM 222765) GNPAT AR RCDP3 (OMIM 600121) AGPS AR RCDP5 (OMIM 616716) PEX5 AR Disorders of post-squalene cholesterol biosynthesis Smith-Lemli-Opitz syndrome DHCR7 AR Skeletal abnormalities incl rhizomelia & polydactyly Significant phenotypic overlap w/MEND syndrome 5 incl ID, facial dysmorphism, multiple congenital abnormalities & genital abnormalities No CDP Antley-Bixler syndrome (see Cytochrome P450 Oxidoreductase Deficiency) POR AR Skeletal abnormalities incl rhizomelia & scoliosis No CDP or skeletal asymmetry Other features incl craniosynostosis, midface hypoplasia, joint contractures, & DD Desmosterolosis (OMIM 602398) DHCR24 AR Skeletal abnormalities incl rhizomelia, joint contractures, & poor growth No CDP or skeletal asymmetry ID Brain & visceral anomalies Lathosterolosis (OMIM 607330) SC5D AR Skeletal abnormalities incl rhizomelia, postaxial polydactyly & spinal abnormalities No CDP or skeletal asymmetry CK (see NSDHL Disorders) NSDHL XL Mild skeletal abnormalities incl scoliosis/kyphosis ID & neuronal migration abnormalities Allelic to CHILD syndrome, but no overlapping features Sterol-C4-methyloxidase-like deficiency (OMIM 616834) MSMO1 ( SC4MOL ) AR Short stature, generalized ichthyosiform dermatitis, & cataracts reported Broader phenotype incl ID, immune dysfunction, & failure to thrive Peroxisome biogenesis disorders Zellweger spectrum disorder PEX1 PEX2 PEX3 PEX5 PEX6 PEX10 PEX11B PEX12 PEX13 PEX14 PEX16 PEX19 PEX26 AR CDP of the patella & long bones Broader phenotype incl congenital malformations, seizures, & liver disease of variable severity AR = autosomal recessive; CDP = chondrodysplasia punctata; CDPX2 = chondrodysplasia punctata 2, X-linked; DD = developmental delay; ID = intellectual disability; MOI = mode of inheritance; XL = X-linked 1.
-
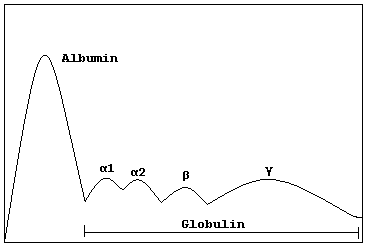
Monoclonal Gammopathy Of Undetermined Significance
Wikipedia
"Rapid onset monoclonal gammopathy in cutaneous lupus erythematosus: interference with complement C3 and C4 measurement". Immunol. Invest . 28 (4): 269–276. doi : 10.3109/08820139909060861 .TNFRSF13B, DNAH11, USP8P1, ULK4, HLA-B, SDC1, ELL2, TOM1, MAG, IL1B, IGH, NCAM1, MYOM2, CDKN2A, MYD88, HGF, CD19, IL6, STOML2, CDKN2B, CXCR4, VEGFA, IL1A, TP53, TNF, MYC, PTPRC, TTR, NRAS, PTPN6, FGF2, SOCS1, ADIPOQ, RASSF1, FOXP3, KRT20, MALAT1, HAS1, H3P10, CKS1B, CTNNB1, CD38, CCND1, MS4A1, ALB, DAPK1, GPNMB, ANP32B, COLEC10, TNFSF13B, PNMA2, NES, LILRB1, SMR3B, LRPPRC, PRSS21, ADAMTS13, PTP4A3, SERPING1, PADI2, KLRK1, BNIP3, DKK1, KDM1A, SEMA3A, CANX, RAD50, PLXNB2, TP73, CD40, VWF, NSD2, XPO1, CD34, CDR3, NR0B2, OFD1, CASK, TNFSF11, CD27, TNKS, TNFSF13, CCND3, MSC, KL, RUNX2, TBPL1, BCL2L1, SETBP1, POT1, ALKBH3, GSTK1, AKT2, MIRLET7E, MIR203A, MIR21, MIR214, MIR27B, MIR34A, MIR340, MIR342, SSX2B, MIR744, CD24, AKT1, MIR1246, KLRC4-KLRK1, PCAT1, LOC102723407, LOC102724971, RICTOR, CLEC12A, CDH1, SLCO6A1, FOXP1, IGHV3OR16-7, IGHV3-69-1, CD274, BCL2, SOST, EVL, WWOX, ALK, EGLN1, NLRP2, SLC12A9, TP73-AS1, AICDA, PREX1, IL21, IGAN1, FOXP2, EGLN3, CD47, TIMP3, DCC, IGL, HSPA5, IGSF3, IFNA2, IGF1, IGF2, IGFBP2, CCN1, CSNK1A1, IGHG3, CR2, TGFBI, COX8A, CCR7, IL6R, IL6ST, CXCL8, IL10, ITGAX, JAG2, JAK2, HRAS, CTLA4, CFH, CYLD, DKC1, EGFR, EPO, EPOR, ESR1, FAP, FCGR3A, DAP, FGFR3, FLT3, FRA16D, MTOR, FUT4, GLI1, GNA12, CXCR3, GPT, GRN, GSTP1, KIT, KRAS, LCK, PTCH1, PARP1, PVR, RALA, RPA1, CCL20, CX3CL1, CDH2, SFRP1, SFRP5, SSX1, SSX2, SSX5, SSX4, STAT3, AURKA, SYK, TRBV20OR9-2, TERF1, TERF2, CDKN1A, PFDN5, LIG3, ENPP1, LPL, LTA, LY9, CCR4, DNAJB9, MET, MGMT, MLH1, MME, MMP2, MRE11, RCBTB2, NBN, CDKN3, NEDD8, NM, PNP, NPC1, CDKN2C, ACTB
-
Plasma Cell Dyscrasias
Wikipedia
This form of MGRS is usually associated with other syndromes like glomerulopathy associated with a monoclonal immunoglobulin or C4 dense deposit disease associated with a monoclonal immunoglobulin. ... Since renal dysfunction usually improves with therapy directed at the underlying plasma cell dyscrasia, MGRS may warrant treatment even when other parameters of plasma cell dyscrasia severity (e.g. low levels of serum monoclonal immunoglobulin and bone marrow plasma cells) suggest the presence of minimal, non-malignant disease. [21] Smoldering multiple myeloma stage [ edit ] Main article: Smoldering multiple myeloma Smoldering multiple myeloma or SMM (also termed smoldering myeloma) is the next stage following MGUS in the spectrum of plasma cell dyscrasias. ... Amyloidosis [ edit ] Main article: Amyloidosis Main article: Light chain deposition disease Amyloidosis is a general term for a protein misfolding syndrome that involves the deposition of a low molecular weight beta-pleated sheet -containing protein in extracellular tissues. ... POEMS is an acronym standing for the characteristic signs or symptoms of the syndrome: P olyneuropathy , O rganomegaly , E ndocrinopathy , P lasma cell disorder (typically, the plasma cell burden is low in POEMS patients), and S kin changes (e.g. hemangioma , hyperpigmentation ). ... Patients with >2 plasmacytomas or symptomatic disseminated disease have been treated with chemotherapy often followed by autologous stem-cell transplantation ; these treatments have been found to reduce symptoms of the disease and lead to long-term partial remissions of disease. [28] [29] The overall survival of POEMS patients who have been treated for their disease is relatively good for a disease occurring in patients with an average age of 50 years; one estimate of median overall survival is 14 years. POEMS patients evaluated to be in low and intermediate risk groups had ≥>85% survival at 10 years; those in the high risk group had a 40% survival over this time period. [30] Cryoglobulinemia [ edit ] Main article: Cryoglobulinemia Cryoglobulins are proteins, principally immunoglobulins , that circulate in the blood, precipitate at temperatures <37 °C (98.6 °F), and re-solubilize upon restoring physiological blood temperatures.
-
Flnb Disorders
GeneReviews
The proportion of autosomal dominant FLNB disorders caused by de novo pathogenic variants is unknown, although the vast majority of lethal FLNB conditions are caused by de novo events. In rare instances, a parent with low-level mosaicism transmits the causative pathogenic variant to an affected offspring. ... Cervical kyphosis was noted in 50%, usually from subluxation or fusion of the bodies of C2, C3, and C4, which was commonly associated with posterior vertebral arch dysraphism (i.e., dysplasia of the vertebral laminae and hypoplasia of the lateral processes of all cervical vertebrae).
- Short Qt Syndrome Wikipedia
-
Serac1 Deficiency
GeneReviews
AUH defect is the only one of the five inborn errors of metabolism with 3-MGA-uria with a distinct biochemical finding: elevated urinary excretion of 3-hydroxyisovaleric acid (3-HIVA). 5. Increased C3- & C4-dicarboxyli-carnitine esters. 6. Wortmann et al [2012a] 7. ... Education of parents/caregivers 1 Dysphagia Feeding therapy Gastrostomy tube placement may be required for persistent feeding issues. Low threshold for clinical feeding eval &/or radiographic swallowing study when showing clinical signs or symptoms of dysphagia Drooling Botulinum toxin injection in salivary glands, extirpation of salivary glands, &/or rerouting of glandular ducts These measures can improve secondary respiratory problems. ... The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices.
-
Varicose Veins
Wikipedia
] Stages [ edit ] The CEAP (Clinical, Etiological, Anatomical, and Pathophysiological) Classification, developed in 1994 by an international ad hoc committee of the American Venous Forum, outlines these stages [24] [25] C0 – no visible or palpable signs of venous disease C1 – telangectasia or reticular veins C2 – varicose veins C2r - recurrent varicose veins C3 – edema C4- changes in skin and subcutaneous tissue due to Chronic Venous Disease C4a – pigmentation or eczema C4b – lipodermatosclerosis or atrophie blanche C4c- Corona phlebectatica C5 – healed venous ulcer C6 – active venous ulcer C6r- recurrent active ulcer Each clinical class is further characterized by a subscript depending upon whether the patient is symptomatic (S) or asymptomatic (A), e.g. ... As of 2015 there is tentative evidence of benefits with a relatively low risk of side effects compared to vein stripping. [34] Ambulatory phlebectomy .VHL, MGP, TIMP1, TNC, DPT, KCNN3, FOXC2, CASZ1, GLG1, GJC2, GP1BB, KIF5A, HLA-A, LBH, FLT4, CNGB3, HIRA, SLC29A3, RREB1, SMAD3, NFATC2, NOTCH3, PIK3CA, PRKAR1B, PIEZO1, SEC24C, RASA1, FIBP, SLC12A2, SLC12A3, TBX1, VEGFC, G6PC3, HDAC7, UFD1, ARVCF, COL3A1, EBF1, CLCNKB, JMJD1C, EPHB4, COMT, LINC02549, LINC01152, ROCR, MMP9, VEGFA, SNCA, BLOC1S2, SYP, SLC17A6, NLRP3, ELN, MMP3, MMP2, MMP1, MTHFR, HIF1A, SLC18A3, TGFB1, STS, CALCA, NPY, GFAP, MCF2L2, TAC1, CALB1, KDR, TIMP2, DBH, ROCK2, CYP4F2, CHST3, CLOCK, HOMER1, RBM14-RBM4, TNFSF11, GNG13, TRPV4, MFAP5, PORCN, VIM, EHMT1, EGLN3, DCLK3, PAPLN, FOXC2-AS1, USH2A, ZGLP1, PPARGC1A, MIR202, AKR1B10, TRPV2, DLL4, SLC17A8, PRLH, EXOSC3, PACSIN1, PYCARD, CBLN4, SLC32A1, TSACC, ADI1, CACYBP, RBM14, KANK2, TP53INP1, BACE2, SLC17A7, HEY2, SYNM, EXOC7, PRRT2, ERC1, PGP, MMRN1, MYOCD, ACHE, TRH, GJA1, F13B, FLT1, FN1, FOS, FOSB, GCG, GJA8, ESR1, GLP1R, GRM1, HCCS, HCRT, HTT, HFE, F2, EPAS1, TIMP3, CALR, ADM, JAG1, AKT1, RHOA, AVP, CALB2, CASP1, DSP, CAV2, CYP4A11, CYP19A1, DES, DLD, DOCK3, ICAM1, ITGB2, ITGB3, CCL2, PRKCA, PRKCB, PTGS2, ACP3, S100A10, S100A12, SELE, JUN, SLC6A3, SLC18A2, SPP1, SST, TH, THBD, PPARG, POU2F1, PLG, PEPD, OPRM1, NOS1, NGFR, NGF, MTR, COX2, MMP13, SMAD2, LIF, LGALS3BP, LAD1, JUND, JUNB, MTCO2P12