-
Immunodeficiency, Partial Combined, With Absence Of Hla Determinants And Beta-2-Microglobulin From Lymphocytes
OMIM
Other gene products coded by chromosome 6 (C2, C4, Chido, Rodgers, Factor B, PGM3 and glyoxalase) were normal.
-
Complement 4 Deficiency
Wikipedia
Complement 4 deficiency Specialty Immunology Complement tests C4 ( C ) FB ( A ) C3 CH50 Conditions · ↓ ↓ ↓ PSG , C3 NeF AA ↓ · ↓ · HAE , C4D · · · ↓ TCPD ↓ · /↓ ↓ ↓ SLE ↑ ↑ ↑ ↑ inflammation Complement 4 deficiency is a genetic condition affecting complement component 4 . [1] It can present with lupus -like symptoms. [2] References [ edit ] ^ Parija (2009).
-
Prolidase Deficiency
GeneReviews
Levels of C3 ranging from 30 to 74 mg/dL (normal: 75-175 mg/dL) have been published [Klar et al 2010], with C4 as low as 9 mg/dL (normal: 20-40 mg/dL) [Cleary et al 1994, Shrinath et al 1997]. ... Although not a universal finding, facial features typically described include prominent forehead, widely spaced eyes, proptosis, depressed nasal bridge, prognathism, thin vermilion of the upper lip, and low anterior and posterior hairlines [Royce & Steinmann 2002, Falik-Zaccai et al 2010, Besio et al 2015]. ... A male age 25 years had bilateral and symmetric synovitis affecting hands, elbows, and knees, a positive rheumatoid factor (516 IU/mL) and homogeneous ANA (1:640), and low C4 and CH50 (0.07 g/L and 61%) [Marotte et al 2010]. ... Of two sibs reported by Klar et al [2010]: A girl age eight years had Raynaud's phenomenon, a positive ANA (1:1280; homogenous pattern), a positive anti ds-DNA >1:160, positive anti-ENA, anti-RNP and anti-Smith; Her brother, age 12 years, had Raynaud's phenomenon, a positive ANA (1:40; homogeneous) and anti ds-DNA (1.6 μg/mL), and low C3 (30-74 mg/dL). An individual with positive ANA and anti-dsDNA titers, as well as low complement levels had no hematologic, renal, or articular problems; although he was given a diagnosis of SLE, he did not fulfill ACR diagnostic criteria [Di Rocco et al 2007]. ... Butbul Aviel et al [2012] reported three individuals with the SLE/prolidase deficiency association: A boy age 4.5 years with a rash consistent with hypertrophic discoid lupus on biopsy, proteinuria and mild hematuria with a renal biopsy consistent with WHO Class IV lupus nephritis, low C3 and C4 levels, positive ANA (1:640; homogeneous pattern), anti ds-DNA, anti-RNP, anti-SM, anti Ro (SS-A) and anti La (SS-B) A girl age 16 years with macroscopic hematuria and proteinuria, with a renal biopsy demonstrating WHO Class IV lupus nephritis; she had low C3 and C4, as well as a highly positive ANA and positive anti ds-DNA titer A woman age 24 years with thrombocytopenia, a positive Coombs test, low C3 and C3 levels, and positive ANA and anti ds-DNA titers Positive ANA, anti-dsDNA, anti-ENA (anti-Ro), anti-Sm and anti-chromatin have been found in individuals with prolidase deficiency even in the absence of clinical findings of SLE [Kurien et al 2013].

-
Muscular Dystrophy-Dystroglycanopathy (Congenital Without Mental Retardation), Type B, 4
OMIM
Mutation in the FKTN gene can also cause a more severe congenital muscular dystrophy-dystroglycanopathy with brain and eye anomalies (type A4; MDDGA4; 253800) and a less severe limb-girdle muscular dystrophy-dystroglycanopathy (type C4; MGDGC4; 611588). Description MDDGB4 is a rare autosomal recessive congenital muscular dystrophy that is part of a group of similar disorders resulting from defective glycosylation of alpha-dystroglycan (DAG1; 128239), collectively known as 'dystroglycanopathies.'
-
Hypoalphalipoproteinemia, Primary, 2
OMIM
The xanthelasmas did not progress after delivery. The woman had absent apoA-I and low HDL-C, but no signs of coronary artery disease or other atherosclerosis. Inheritance Borecki et al. (1986) studied 16 kindreds ascertained through probands clinically determined to have primary hypoalphalipoproteinemia characterized by low HDL cholesterol but otherwise normal blood lipids. ... As a result, the apoA-I protein is not synthesized or secreted, leading to absent apoA-I in plasma and very low levels of HDL-C. Missense mutations in the apoA-I gene, which are almost always heterozygous, affect the structure of the apoA-I protein, often leading to impaired function and/or increased metabolism and low levels of apoA-I and HDL-C (Rader and deGoma, 2012). ... The frequency of hypoalphalipoproteinemia due to a mutant APOA1 gene was estimated at 6% in subjects with low HDL-c levels and 0.3% in the Japanese population generally. ... In a 67-year-old Japanese male with corneal opacities, coronary artery disease, and low apoA-I and HDL-C levels, Huang et al. (1998) identified a homozygous mutation in the APOA1 gene (V156E; 107680.0022).
-
Complement Factor I Deficiency
OMIM
She had no circulating factor I and low C3 (approximately 30% of normal controls). ... Both the proband and his sister had no detectable circulating factor I or factor B, and a very low complement C3 level, most of which was in the form of C3B. ... Renal biopsy showed focal segmental glomerulonephritis with glomerular deposits of immunoglobulins and complement C3 and C4 fragments. In addition, there appeared to be depletion of complement receptor I (CR1; 120620). ... Those with complete deficiency had recurrent respiratory infections, skin infections, and meningitis; 1 died from sepsis. All had low serum C3, low factor H, and undetectable factor I activity.

-
Type I Hypersensitivity
Wikipedia
The immediate hypersensitivity reaction occurs minutes after exposure and includes release of vasoactive amines and lipid mediators, whereas the late-phase reaction occurs 2–4 hours after exposure and includes the release of cytokines . [4] List of a few mediators released by mast cells in type 1 hypersensitivity and their actions Vasodilation and increased permeability Histamine PAF Leukotriene C4 , D4 , and E4 Prostaglandin D2 Neutral proteases Smooth muscle spasm Histamine PAF Leukotriene C4, D4, and E4 Prostaglandin Leukocyte extravasation Cytokines (e.g. chemokines and TNF ) Leukotriene B4 Chemotactic factors for neutrophils and eosinophils Unless otherwise specified, the reference for this table is: [5] The reaction may be either local or systemic.

-
Odontoid Hypoplasia
OMIM
He had progressive neurologic symptoms and underwent additional surgery for occipital to C4 fusion. He has quadriparesis, more severe in the upper extremities, and is not able to walk or stand.
-
C3 Glomerulopathy
GeneReviews
Individuals with C3G typically present with hematuria, proteinuria, hematuria and proteinuria, acute nephritic syndrome or nephrotic syndrome, and low levels of the complement component C3. ... Suggestive Findings C3G should be suspected in individuals of all ages who present with one of the following: Hematuria Proteinuria Hematuria and proteinuria Acute nephritic syndrome Nephrotic syndrome Persistent hypocomplementemia (low serum levels of complement component C3) Establishing the Diagnosis The diagnosis of C3G is established in a proband with typical findings on renal biopsy . ... Individuals with C3G typically present with one of the following findings: Hematuria Proteinuria Hematuria and proteinuria Acute nephritic syndrome Nephrotic syndrome Hypocomplementemia. Individuals with C3G have low levels of complement component C3. ... Acquired drivers of disease include autoantibodies such as C3 nephritic factors (C3NeFs), C4 nephritic factors (C4NeFs), C5 nephritic factors (C5NeFs), factor H autoantibodies (FHAA), and factor B autoantibodies (FBAA). ... Stone et al [1999], Mullins et al [2001], Sohn et al [2015], Hulleman [2016], Vaclavik & Munier [2016] Management Evaluations Following Initial Diagnosis To establish the extent of disease and needs in an individual diagnosed with C3G, the following evaluations are recommended if they have not already been completed: Evaluate the complement system by measuring serum/plasma concentrations of C3, C3c, C3d, C4, C5, fB, Ba, Bb, fH, fI, properdin, and s(C5b-9).
-
Aspirin Exacerbated Respiratory Disease
Wikipedia
One confounding factor in the study that showed a benefit from avoidance of dietary salicylates is that a low salicylate diet involves eliminating wine and beer. ... In contrast to aspirin, dietary salicylates are not acetylated and therefore do not block cyclooxygenases and hence, there is no rationale why a low salicylate diet would be beneficial for AERD patients. [40] [ unreliable medical source ] A diet low in omega-6 oils (precursors of arachidonic acid), and high in omega-3 oils may also be of benefit. ... PMID 12421891 . ^ Cowburn AS, Sladek K, Soja J, Adamek L, Nizankowska E, Szczeklik A, Lam BK, Penrose JF, Austen FK, Holgate ST, Sampson AP (February 1998). "Overexpression of leukotriene C4 synthase in bronchial biopsies from patients with aspirin-intolerant asthma" . ... "A novel treatment adjunct for aspirin exacerbated respiratory disease: the low-salicylate diet: a multicenter randomized control crossover trial". ... PMC 6455762 . PMID 29747986 . ^ "A Low Salicylate Diet for AERD (Samter's Triad)" .

-
Leiner's Disease
Wikipedia
Leiner's disease Specialty Dermatology Leiner's disease is a systemic disease, a skin disorder and extends to erythroderma, typically diagnosed in early infancy. [1] Leiner’s disease is characterized by a long-lasting seborrhea dermatitis associated with increased likelihood to infection. [2] Other characterizations found on newborns with Leiner’s disease are a patch or a large patch of red skin normally on the bottom and spreads to the rest of the body. [2] This disease is also listed as a "rare disease", meaning that a small percent of the population, fewer than 200,000 people in the United States, will have this disorder. [2] Contents 1 Symptoms 2 Cause 3 Diagnosis 4 Treatment 5 Epidemiology 6 See also 7 References 8 External links Symptoms [ edit ] Symptoms include severe seborrheic dermatitis of the scalp, severe diarrhea, recurrent local and systemic infection, central nervous system problems, and failure to thrive . [3] Other symptoms also include scaling on the trunk and limbs, red patches of skin on parts of the body that bend, fevers, reduced blood protein levels, thick red skin patches, peeling of the skin, itching, corneal ulcers. wasting of the lymph nodes, underdeveloped lymphatics, anemia, wasting, and nervous system deficiency. [2] The disease may then spread to the rest of the epidermis with the appearance of crusty, dry, moist or greasy scaling on the scalp. [1] Scaling could also appear behind the ears, nose or eyebrows, or around the mouth; peeling of the skin may also happen in these areas. [1] If left untreated, the skin infections will cause loss of protein or electrolytes. [1] Leiner’s Disease may also be accompanied by a systemic reaction that is most evident in its gastrointestinal manifestation. [4] It is caused by a deficit of the complement protein , C5 ; however, case reports have described it in relation to deficits in either C3 or C4 . [5] Cause [ edit ] The exact cause of Leiner disease remains unknown but biotin deficiency linked to Complement component 5 (a protein ) coded by the C5 gene plays a role.
-
Flexion Teardrop Fracture
Wikipedia
Flexion teardrop fractures usually involve instability in all elements of the spine at the injured level, commonly occur at the C4-C7 vertebra, and have a high association with spinal cord injury (in particular anterior cord syndrome ).

-
Adrenal Hyperplasia, Congenital, Due To 21-Hydroxylase Deficiency
OMIM
They concluded that the pathophysiologic significance of this finding in the presence of 1 normal CYP21B gene seems to be low, suggesting that 21-hydroxylase deficiency is not a major predisposing factor for adrenal tumor formation. ... Those who were heterosexually active nevertheless appeared to have low fertility. Among the 25 SV women who reported both adequate vaginal reconstruction and heterosexual activity, the fertility rate was 60%. ... Patients with the HLA-Bw47 antigen invariably show simultaneous deficiencies of 21-hydroxylase activity and the C4A (Rodgers) form of C4. The HLA-Bw47(w4) antigen is very similar serologically and otherwise to the more common antigen HLA-B13(w4). ... The patient's lack of response to treatment with cortisone acetate was caused by a low conversion of cortisone to cortisol, assumed to be secondary to low 11-beta-hydroxysteroid dehydrogenase activity. ... Falhammar et al. (2007) concluded that women with CAH have low BMD and increased fracture risk, and suggested that BMD should be monitored, adequate prophylaxis and treatment instituted, and glucocorticoid doses optimized from puberty.CYP17A1, HSD3B2, CYP21A2, POR, PDE8B, PRKAR1A, AVPR2, HTR4, AVPR1A, CYP11B1, STAR, CYP11B2, PDE11A, KCNJ5, PRKACA, USP8, CDH23, CYP21A1P, CYP2B6, POMC, NR3C1, GML, AIRE, CACNA1D, TBC1D24, TNXB, AR, HSD3B1, CHST3, CYP19A1, GLO1, HLA-A, CYP4F3, NR0B1, MC2R, HSD11B2, TNXA, SRY, REN, GIP, HLA-B, GH1, CYP2C19, CRH, RNU1-4, TWIST1, SDHD, TP53, PPIG, SCO2, VWF, TGM5, SULT2A1, ARMC5, COASY, CGB5, CGB8, RBM45, OTOA, EYS, ACADVL, SDHB, PROP1, ATP7B, AVP, AZF1, BCL2, C4B, CGA, CGB3, CRYGD, CYP3A7, CYP2B7P, CYP11A1, ENPEP, FH, GRB7, HADHA, HLA-DRB1, HTC2, TNC, LEP, MDH2, MEN1, SERPINA1, PRKACB, LOC110673972
-
Tetraplegia
Wikipedia
Tetraplegia is defined in many ways; C1–C4 usually affects arm movement more so than a C5–C7 injury; however, all tetraplegics have or have had some kind of finger dysfunction. ... In high-level cervical injuries, total paralysis from the neck can result. High-level tetraplegics (C4 and higher) will likely need constant care and assistance in activities of daily living , such as getting dressed, eating and bowel and bladder care. Low-level tetraplegics (C5 to C7) can often live independently. [ citation needed ] Even with "complete" injuries, in some rare cases, through intensive rehabilitation, slight movement can be regained through "rewiring" neural connections , as in the case of actor Christopher Reeve . [6] In the case of cerebral palsy , which is caused by damage to the motor cortex either before, during (10%), or after birth, some people with tetraplegia are gradually able to learn to stand or walk through physical therapy. [ citation needed ] Quadriplegics can improve muscle strength by performing resistance training at least three times per week. Combining resistance training with proper nutrition intake can greatly reduce co-morbidities such as obesity and type 2 diabetes. [7] Epidemiology [ edit ] See also: List of people with quadriplegia There are an estimated 17,700 spinal cord injuries each year in the United States; the total number of people affected by spinal cord injuries is estimated to be approximately 290,000 people. [8] In the US, spinal cord injuries alone cost approximately US$40.5 billion each year, which is a 317 percent increase from costs estimated in 1998 ($9.7 billion). [9] The estimated lifetime costs for a 25-year-old in 2018 is $3.6 million when affected by low tetraplegia and $4.9 million when affected by high tetraplegia. [8] In 2009, it was estimated that the lifetime care of a 25-year-old rendered with low tetraplegia was about $1.7 million, and $3.1 million with high tetraplegia. [10] There are about 1,000 people affected each year in the UK (~1 in 60,000—assuming a population of 60 million).TBCD, PMP22, PRF1, ALS2, AIFM1, TXNRD2, PNKP, NNT, STAR, TRAPPC2L, SLC6A3, MRAP, KCNT1, ALDH18A1, PRPS1, ARSA, BCAP31, TFG, IRF2BPL, GABRA3, CACNA1S, KCNJ18, DPYD, NHLRC2, NADK2, FGFR1, ARX, KRAS, MC2R, MECP2, IS1, TP63, AR, CENPJ, SOST, PYCARD, REM1, ALDH3A2, CLOCK, RANBP2, COL4A1, F5, IGFALS, IL4, MTHFR, FXYD1, PLP1, RPE, LDB1, SCT, SOD1, APOE, TNF, TYMS, UCP2, EOS, PERCC1
-
Spondyloepiphyseal Dysplasia With Congenital Joint Dislocations
OMIM
On x-ray, in addition to previously described findings, he had narrow disc spaces at C3-C4 and C4-C5, subluxation of C4-C5, marked lumbar lordosis and dorsal kyphosis, herniation of the discs of the dorsolumbar spine, irregular vertebral endplates of the vertebrae of the thoracolumbar spine, lumbar spina bifida occulta, abnormal carpal bones with extra-carpal bones on the left hand, abnormal tarsal bones, accessory medial cuneiforms, slightly flared metaphyses, and abnormal epiphyses.
-
Nephrotic Syndrome, Type 7
OMIM
At onset, all had proteinuria and most had low serum albumin. In 1 family, the proband presented with nephrotic syndrome and renal insufficiency at age 2 years and died of meningitis a year later. ... Serum complement components C3 and C4 (see 120810) were normal in all individuals; serum CFH (134370) was not measured. ... INHERITANCE - Autosomal recessive GENITOURINARY Kidneys - Hemolytic uremic syndrome (in some patients) - Acute renal failure - Nephrotic syndrome - Proteinuria - Membranoproliferative glomerulonephritis seen on biopsy - Thickening of the glomerular basement membrane - Splitting of the basement membrane - Focal capillary obliteration - Mesangial cell proliferation - Effacement of podocyte foot processes - Patchy deposition of IgG and IgM - Subendothelial deposits - Swelling of endothelial cells - Chronic thrombotic microangiopathy - End-stage renal failure (in some patients) HEMATOLOGY - Hemolytic anemia (in some patients) - Thrombocytopenia (in some patients) LABORATORY ABNORMALITIES - Low serum albumin - Normal serum complement levels MISCELLANEOUS - Onset usually in the first decade (range 0.8 to 5 years) - Atypical hemolytic-uremic syndrome shows onset in first 12 months - Progressive disorder - Some patients may show response to immunosuppressive agents - Some patients do not reach end-stage renal failure MOLECULAR BASIS - Caused by mutation in the diacylglycerol kinase, epsilon, 64-kD gene (DGKE, 601440.0001 ) ▲ Close
-
Spinal Cord Injury
Wikipedia
In younger people, it most commonly results from neck flexion. [26] The most common causes are falls and vehicle accidents; however other possible causes include spinal stenosis and impingement on the spinal cord by a tumor or vertebral disk. [27] Anterior cord syndrome [ edit ] Anterior cord syndrome , due to damage to the front portion of the spinal cord or reduction in the blood supply from the anterior spinal artery , can be caused by fractures or dislocations of vertebrae or herniated disks. [25] Below the level of injury, motor function, pain sensation, and temperature sensation are lost, while sense of touch and proprioception (sense of position in space) remain intact. [28] [26] These differences are due to the relative locations of the spinal tracts responsible for each type of function. [25] Brown-Séquard syndrome [ edit ] Brown-Séquard syndrome occurs when the spinal cord is injured on one side much more than the other. [29] It is rare for the spinal cord to be truly hemisected (severed on one side), but partial lesions due to penetrating wounds (such as gunshot or knife wounds) or fractured vertebrae or tumors are common. [30] On the ipsilateral side of the injury (same side), the body loses motor function, proprioception , and senses of vibration and touch. [29] On the contralateral (opposite side) of the injury, there is a loss of pain and temperature sensations. [27] [29] Posterior cord syndrome [ edit ] Posterior cord syndrome , in which just the dorsal columns of the spinal cord are affected, is usually seen in cases of chronic myelopathy but can also occur with infarction of the posterior spinal artery . [31] This rare syndrome causes the loss of proprioception and sense of vibration below the level of injury [26] while motor function and sensation of pain, temperature, and touch remain intact. [32] Usually posterior cord injuries result from insults like disease or vitamin deficiency rather than trauma. [33] Tabes dorsalis , due to injury to the posterior part of the spinal cord caused by syphilis, results in loss of touch and proprioceptive sensation. [34] Conus medullaris and cauda equina syndromes [ edit ] Conus medullaris syndrome is an injury to the end of the spinal cord, located at about the T12–L2 vertebrae in adults. [29] This region contains the S4–S5 spinal segments, responsible for bowel, bladder, and some sexual functions , so these can be disrupted in this type of injury. [29] In addition, sensation and the Achilles reflex can be disrupted. [29] Causes include tumors , physical trauma, and ischemia . [35] Cauda equina syndrome (CES) results from a lesion below the level at which the spinal cord splits into the cauda equina , [33] at levels L2–S5 below the conus medullaris. [36] Thus it is not a true spinal cord syndrome since it is nerve roots that are damaged and not the cord itself; however, it is common for several of these nerves to be damaged at the same time due to their proximity. [35] CES can occur by itself or alongside conus medullaris syndrome. [36] It can cause low back pain, weakness or paralysis in the lower limbs, loss of sensation, bowel and bladder dysfunction, and loss of reflexes. [36] Unlike in conus medullaris syndrome, symptoms often occur on only one side of the body. [35] The cause is often compression, e.g. by a ruptured intervertebral disk or tumor. [35] Since the nerves damaged in CES are actually peripheral nerves because they have already branched off from the spinal cord, the injury has better prognosis for recovery of function: the peripheral nervous system has a greater capacity for healing than the central nervous system . [36] Signs and symptoms [ edit ] Actions of the spinal nerves Level Motor Function C1 – C6 Neck flexors C1 – T1 Neck extensors C3 , C4 , C5 Supply diaphragm (mostly C4 ) C5 , C6 Move shoulder , raise arm ( deltoid ); flex elbow ( biceps ) C6 externally rotate ( supinate ) the arm C6 , C7 Extend elbow and wrist ( triceps and wrist extensors ); pronate wrist C7 , T1 Flex wrist; supply small muscles of the hand T1 – T6 Intercostals and trunk above the waist T7 – L1 Abdominal muscles L1 – L4 Flex thigh L2 , L3 , L4 Adduct thigh; Extend leg at the knee ( quadriceps femoris ) L4 , L5 , S1 abduct thigh; Flex leg at the knee ( hamstrings ); Dorsiflex foot ( tibialis anterior ); Extend toes L5 , S1 , S2 Extend leg at the hip ( gluteus maximus ); Plantar flex foot and flex toes Further information: Dermatome (anatomy) Signs (observed by a clinician) and symptoms (experienced by a patient) vary depending on where the spine is injured and the extent of the injury. ... For example, problems with body temperature regulation mostly occur in injuries at T8 and above. [42] Another serious complication that can result from lesions above T6 is neurogenic shock , which results from an interruption in output from the sympathetic nervous system responsible for maintaining muscle tone in the blood vessels. [5] [45] Without the sympathetic input, the vessels relax and dilate. [5] [45] Neurogenic shock presents with dangerously low blood pressure, low heart rate , and blood pooling in the limbs—which results in insufficient blood flow to the spinal cord and potentially further damage to it. [46] Cervical [ edit ] Spinal cord injuries at the cervical (neck) level result in full or partial tetraplegia (also called quadriplegia). [24] Depending on the specific location and severity of trauma, limited function may be retained. Additional symptoms of cervical injuries include low heart rate , low blood pressure , problems regulating body temperature , and breathing dysfunction. [47] If the injury is high enough in the neck to impair the muscles involved in breathing, the person may not be able to breathe without the help of an endotracheal tube and mechanical ventilator. [9] Function after complete cervical spinal cord injury [48] Level Motor Function Respiratory function C1–C4 Full paralysis of the limbs Cannot breathe without mechanical ventilation C5 Paralysis of the wrists, hands, and triceps Difficulty coughing, may need help clearing secretions C6 Paralysis of the wrist flexors, triceps, and hands C7–C8 Some hand muscle weakness, difficulty grasping and releasing Complications [ edit ] Complications of spinal cord injuries include pulmonary edema , respiratory failure , neurogenic shock , and paralysis below the injury site.
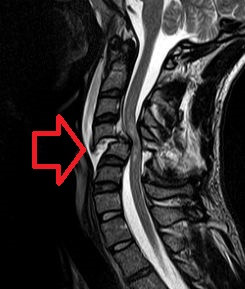
- Renal Failure, Progressive, With Hypertension OMIM
-
Angioedema, Hereditary, Type I
OMIM
-
Search Hereditary Angioedema case abstracts
Detailed laboratory investigations revealed decreased serum levels of several complement components, including C2, C4, C1q, and C1INH. Nerve conduction studies indicated a sensorimotor axonal peripheral neuropathy. ... In 78%, both antigen levels and functional activity of C1 esterase inhibitor were low (HANE type I). Quastel et al. (1983) studied the catabolism of C1 inhibitor in HANE I. ... In this family, homozygosity correlated with low C1 inhibitor levels and severe HANE. ... She had been treated with an attenuated androgen in low dose (danazol and then amicar), which raised her C1 esterase inhibitor level and controlled her symptoms. ... INHERITANCE - Autosomal dominant - Autosomal recessive RESPIRATORY Nasopharynx - Pharyngeal edema Larynx - Laryngeal edema ABDOMEN - Abdominal pain Gastrointestinal - Intestinal edema - Diarrhea - Vomiting SKIN, NAILS, & HAIR Skin - Erythema marginatum MUSCLE, SOFT TISSUES - Episodic, nonpruritic, nonurticarial, nonpitting edema NEUROLOGIC Peripheral Nervous System - Peripheral axonal neuropathy, distal, vasculitic - Sural nerve biopsy shows axonal degeneration - Impaired sensation of all modalities, distal LABORATORY ABNORMALITIES - C1 esterase inhibitor deficiency - Low level of C4 and C2 MISCELLANEOUS - Symptoms typically begin in childhood - Prevalence estimated at 1 in 50,000 - Highly variable frequency and severity of attacks - Trauma, anxiety, and/or stress can precipitate or aggravate edema - Laryngeal edema can result in asphyxiation - Associated with increased frequency of autoimmune diseases - Several patients with homozygous C1NH mutations have been reported (see 606860.0013 ) MOLECULAR BASIS - Caused by mutation in the C1 esterase inhibitor gene (C1NH, 106100.0001 ) ▲ Close
-
Spondylolisthesis
Wikipedia
S2CID 25145462 . ^ a b "Adult Spondylolisthesis in the Low Back" . American Academy of Orthopaedic Surgeons . ... "Determination of spondylolisthesis in low back pain by clinical evaluation". ... "Diagnostic imaging for low back pain: advice for high-value health care from the American College of Physicians" . ... S2CID 31602395 . ^ Casazza, Brian A. (2012-02-15). "Diagnosis and Treatment of Acute Low Back Pain" . American Family Physician . 85 (4): 343–350. ... "Management of symptomatic degenerative low-grade lumbar spondylolisthesis" .BMP2, AGA, RUNX2, CTSK, ZNF469, FBN1, PABPN1, IDUA, TGFBR2, SMAD3, MAN2B1, AMH, PLF, RANBP2, MMP3, SMS, LIF, TRP-AGG2-6, TRP-TGG3-1, TRPC3, FTH1, GDF5, PLA2G15, ACSS2, DCT, ACCS, RSS, ROM1







