-
Simple Cryoglobulinemia
Orphanet
Patients with simple cryoglobulinemia lack the typical vasculitic manifestations and serological findings (RF positivity and low complement C4) that characterize MC patients.
-
Vertebral Anomalies And Variable Endocrine And T-Cell Dysfunction
OMIM
The son and daughter had thymus aplasia or hypoplasia, with absent or low T cell numbers, whereas their mother had low naive T cells. ... Endocrine anomalies in this family included autoimmune hypothyroidism in the son, hypoparathyroidism in the daughter, and low-normal PTH in the mother. The son showed average intelligence with attention-deficit/hyperactivity disorder and autistic behaviors, whereas the daughter showed mild developmental delay; their mother had normal intelligence. ... INHERITANCE - Autosomal dominant GROWTH Height - Short stature HEAD & NECK Head - Brachycephaly Face - Triangular face - Low anterior hairline - Glabellar hemangioma Ears - Low-set ears - Cupped ears - Overfolded helices Eyes - Hypertelorism - Epicanthal folds - Corectopia Nose - Depressed nasal bridge - Depressed nasal tip - Broad nasal tip Mouth - Cleft palate - High-arched narrow palate - Cleft lip Neck - Short neck - Webbed neck CARDIOVASCULAR Heart - Double-outlet right ventricle - Pulmonary valve stenosis - Atrial septal defect Vascular - Patent ductus arteriosus CHEST Ribs Sternum Clavicles & Scapulae - Sprengel deformity - Fusion of 4th and 5th ribs SKELETAL Spine - Klippel-Feil anomaly (fusion C2-C4) - Congenital fusions of thoracic spine - Open laminae posteriorly - Hemivertebrae at T10-T11 - Scoliosis - Kyphosis Hands - Camptodactyly 3rd and 4th digits NEUROLOGIC Central Nervous System - Developmental delay, mild - Attention-deficit hyperactivity disorder (ADHD) Behavioral Psychiatric Manifestations - Autistic behaviors ENDOCRINE FEATURES - Hashimoto thyroiditis - Hypoparathyroidism - Borderline low parathyroid hormone (PTH) - Growth hormone deficiency IMMUNOLOGY - Thymus aplasia or hypoplasia - Low or no functional T cells - Abnormal B cells - Very low naive T cells (CD4 and CD8) MISCELLANEOUS - Variable features may be present MOLECULAR BASIS - Caused by mutation in the T-box 2 gene (TBX2, 600747.0001 ) ▲ Close
-
Spondylocostal Dysostosis 6, Autosomal Recessive
OMIM
The proband had failure of formation of the posterior elements of C1 to C4 with descent of the occipital bone, causing spinal canal stenosis and spinal cord compression. ... His brother had deficiency of the posterior elements of C1 to C3, left hemivertebrae at C4 and T9, and a right hemivertebra at T4, causing marked cervical kyphosis at the C2/C3 level with associated cord compression.

-
Ethylmalonic Encephalopathy
Orphanet
In addition to increased excretion of EMA, methylsuccinic acid and C4-C6-acylglycines (isobutyryl-, isovaleryl-, 2-methylbutyryl-, hexanoylglycine) may also be found in small, but elevated, amounts in the urine. Blood levels of C4-C6-carnitines (butyryl-, isobutyryl-, isovaleryl- and hexanoylcarnitine) may be elevated.ETHE1, TRIM8, TNK2, ZNF575, BDNF, TNF, OXR1, BCL11A, HBS1L, POSTN, BECN1, ABCB11, TRPV1, ALDH3A2, ANXA5, MYB, MUC1, IL6, GRM5, ETFA, TYMP, DNM1, ABCC2, CALB2, BNIP3, SOD2

-
Hereditary Angioedema
Orphanet
-
Search Hereditary Angioedema case abstracts
Diagnostic methods Diagnosis of HAE types 1 and 2 relies on measurement of C4 concentrations and on quantitative and functional analysis of C1-INH. Diagnosis of HAE type 3 revolves around recognition of the clinical picture; C4 and C1-INH levels are normal. Analysis for mutations in the F12 gene may be proposed but are present in only 15% of patients.F12, SERPING1, KNG1, PLG, GRK6, KLKB1, SPINK6, SERPINA4, ANGPT1, ACE, SMPX, POMC, SERPINF2, INA, RBPJ, SERPINF1, CSHL1, CRP, BDKRB2, C2, C1S, CPQ, MBL3P, MASP2, ABCB6, SSB, VCAM1, TNF, SMUG1, TGM2, IL23A, C4orf3, LINC01194, TGFB3, TFPI, A2M, RO60, TRIM21, ABO, ADM, AHSG, ALB, C4B, F3, NR3C1, HSP90AA1, IL17A, MBL2, NHS, SERPINB2, AAVS1, PLAT, REN, TLX1NB

-
Schizophrenia 3
OMIM
For discussion of the contribution of the C4 locus on chromosome 6p21 to schizophrenia risk, see MOLECULAR GENETICS. ... In postmortem human adult brain samples, Sekar et al. (2016) found that C4 RNA expression was directly proportional to C4 copy number; that expression levels of C4A were 2 to 3 times greater than levels for C4B, even after controlling for relative copy number; and that copy number of the C4-HERV sequence increased the ratio of C4A to C4B expression. Sekar et al. (2016) characterized the structural variation of human C4 genes (C4A and C4B) and evaluated association to over 7,000 SNPs across the extended MHC locus (chr6:25-34 Mb), C4 structural alleles, and HLA sequence polymorphisms in 28,799 schizophrenia cases and 35,986 controls. ... The other peak of association centered at the C4 locus, where schizophrenia associated most strongly with the genetic predictor of C4A expression levels (p = 3.6 X 10(-24)). ... Studies in mice demonstrated defects in synaptic remodeling in C4-deficient mice. Sekar et al. (2016) concluded that association of schizophrenia with the MHC locus on chromosome 6p21.3 involves many common, structurally distinct C4 alleles that affect expression of C4A and C4B in the brain, with each allele associated with schizophrenia risk in proportion to its effect on C4A expression.
-
Human Papillomavirus Type 18 Integration Site 1
OMIM
In 2 cervical carcinoma cell lines, HeLa and C4-I, Durst et al. (1987) found that HPV18 DNA was integrated into chromosome 8, 5-prime to the MYC gene (190080).
-
Immunodeficiency Due To A Classical Component Pathway Complement Deficiency
Orphanet
Immunodeficiency due to a classical component pathway complement deficiency is a primary immunodeficiency due to a deficiency in either complement components C1q, C1r, C1s, C2 or C4 characterized by increased susceptibility to bacterial infections, particularly with encapsulated bacteria, and increased risk for autoimmune disease.
-
Serum Sickness-Like Reaction
Wikipedia
Laboratory abnormalities include normal or mild decreases in serum C3, C4, and CH50 levels, and mild proteinuria.1,3-5 In contrast to true serum sickness, renal and hepatic involvement is rare. Significant decreases in serum C3, C4, and CH50, reported in the literature for true serum sickness, are rarely described in serum sickness–like reaction.
-
Microcephaly, Congenital Cataract, And Psoriasiform Dermatitis
OMIM
Serum lipid profile showed persistently low total cholesterol, HDL, and LDL, with normal triglycerides and VLDL. ... Experiments with culture medium were consistent with a block at the step of sterol-C4-methyl oxidase (607545) in the cholesterol synthesis pathway. ... INHERITANCE - Autosomal recessive GROWTH Height - Short stature Weight - Low weight HEAD & NECK Head - Microcephaly Eyes - Cataract, congenital - Blepharitis GENITOURINARY Internal Genitalia (Female) - Delayed puberty SKELETAL - Delayed bone age Limbs - Arthralgias - Joint contractures, especially of lower extremities SKIN, NAILS, & HAIR Skin - Ichthyosiform erythroderma sparing the palms Skin Histology - Psoriasiform hyperplasia - Dilated capillaries in dermal papillae - Neutrophils in epidermis - Neutrophils in stratum corneum - CD68-negative lipid-containing foamy cells in dermis Hair - Fine, lusterless hair NEUROLOGIC Central Nervous System - Mental retardation, mild METABOLIC FEATURES - Low total cholesterol - Low HDL - Low LDL ENDOCRINE FEATURES - Delayed puberty IMMUNOLOGY - Markedly elevated IgE - Markedly elevated IgA - Increase in CD16+ activated granulocytes - Increase in TLR2+ ( 603028 )/TLR4- ( 603030 ) granulocytes - Decrease in granulocyte-specific CD16B ( 610665 ) isoform - Elevated IL6 ( 147620 ) - Elevated IL8 ( 146930 ) - Increase in proinflammatory cytokines - Increase in proinflammatory chemokines MISCELLANEOUS - Heterozygotes exhibit subclinical metabolic and immunologic abnormalities - Based on report of 2 unrelated patients (last curated February 2016) MOLECULAR BASIS - Caused by mutation in the methylsterol monooxygenase 1 gene (MSMO1, 607545.0001 ) ▲ Close
-
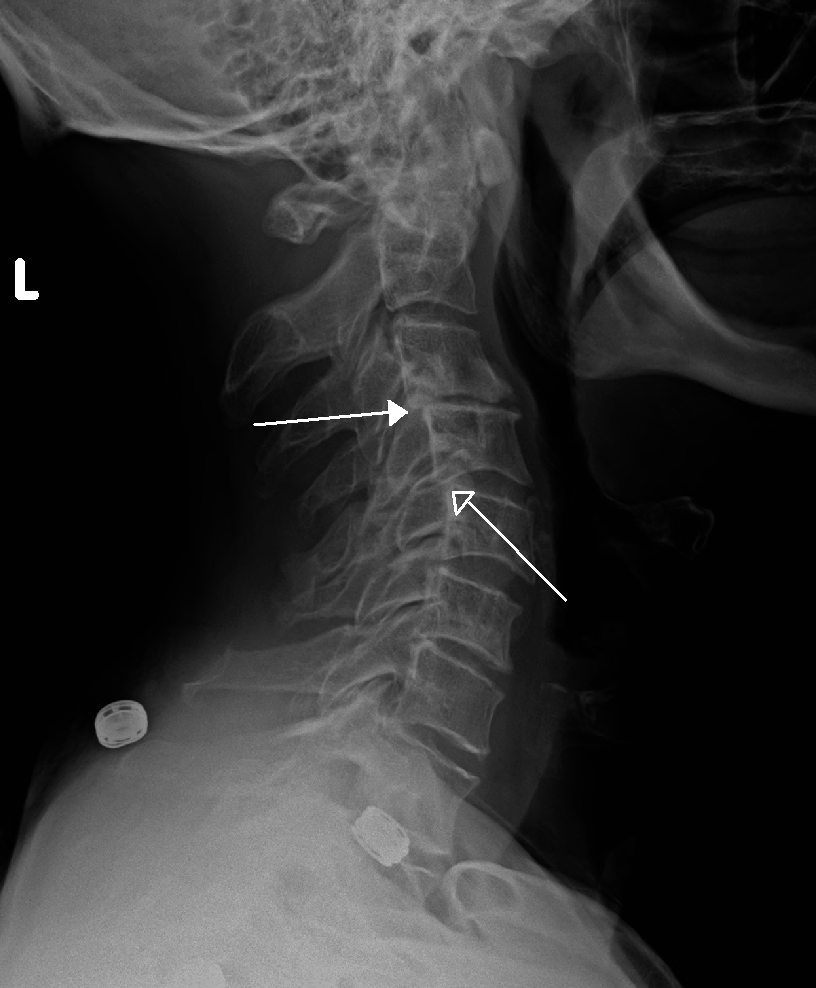
Retrolisthesis
Wikipedia
Retrolisthesis Grade 1 retrolistheses of C3 on C4 and C4 on C5 Specialty Orthopedics A retrolisthesis is a posterior displacement of one vertebral body with respect to the subjacent vertebra to a degree less than a luxation (dislocation) . ... British Volume, Vol 88-B, Issue SUPP_III, 450. v t e Spinal disease Deforming Spinal curvature Kyphosis Lordosis Scoliosis Other Scheuermann's disease Torticollis Spondylopathy inflammatory Spondylitis Ankylosing spondylitis Sacroiliitis Discitis Spondylodiscitis Pott disease non inflammatory Spondylosis Spondylolysis Spondylolisthesis Retrolisthesis Spinal stenosis Facet syndrome Back pain Neck pain Upper back pain Low back pain Coccydynia Sciatica Radiculopathy Intervertebral disc disorder Schmorl's nodes Degenerative disc disease Spinal disc herniation Facet joint arthrosis
-
Unilateral Nevoid Telangiectasia
Wikipedia
Please help improve it to make it understandable to non-experts , without removing the technical details. ( November 2018 ) ( Learn how and when to remove this template message ) Unilateral nevoid telangiectasia Specialty Dermatology Unilateral nevoid telangiectasia presents with fine, threadlike telangiectases , developing in a unilateral, sometimes dermatomal, distribution, with the areas most often involved being the trigeminal and C3 and C4 or adjacent areas. [1] : 589,844 [2] See also [ edit ] Skin lesion List of cutaneous conditions References [ edit ] ^ James, William; Berger, Timothy; Elston, Dirk (2005).
-
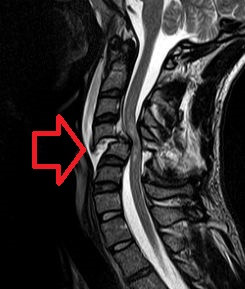
Neurogenic Shock
Wikipedia
Neurogenic shock Cervical spine MRI of a patient with SCI : C4 fracture and dislocation, spinal cord compression Specialty Neurology Neurogenic shock is a distributive type of shock resulting in low blood pressure , occasionally with a slowed heart rate , that is attributed to the disruption of the autonomic pathways within the spinal cord. It can occur after damage to the central nervous system , such as spinal cord injury and traumatic brain injury . Low blood pressure occurs due to decreased systemic vascular resistance as a result of lacking sympathetic tone which in turn causes pools of blood staying within the extremities and not being redirected to the core body. ... Neurogenic shock's presentation includes: [5] [6] - warm and pink skin - labored breathing - low blood pressure - dizziness - anxiety - history of trauma to head or upper spine. - if the injury is to the head or neck, hoarseness or difficulty swallowing may occur. ... External links [ edit ] Classification D ICD - 10 : R57.8 ICD - 9-CM : 785 MeSH : D012769 v t e Shock Distributive Septic shock Neurogenic shock Anaphylactic shock Toxic shock syndrome Obstructive Abdominal compartment syndrome Low volume Hemorrhage Hypovolemia Osmotic shock Other Spinal shock Cryptic shock Vasodilatory shock
-
Multiple Acyl-Coa Dehydrogenase Deficiency
Orphanet
They usually have dysplastic kidneys with multiple cysts and may also have facial dysmorphism (low-set ears, high forehead, hypertelorism and hypoplastic midface), rocker-bottom feet and anomalies of external genitalia. ... Blood acylcarnitines show increased C4-C18 species although patients may be severely carnitine depleted, which may limit the degree of these abnormalities.
-
Glutathionuria
Wikipedia
External links [ edit ] Classification D OMIM : 231950 MeSH : C536836 v t e Eicosanoid metabolism disorders Prostanoids PTGIS ( Essential hypertension ) TBSAX1 ( Ghosal hematodiaphyseal dysplasia ) Leukotrienes LTC4s ( Leukotriene C4 synthase deficiency ) GGT1 ( Glutathionuria ) Other/ungrouped HPGD ( Primary hypertrophic osteoathropathy ) This medical sign article is a stub .
-
Complement Component 2 Deficiency
OMIM
Since two were homozygous HLA-A10, Bw18 and the third was a (A10, B11) (A2, B12.2) heterozygote, the authors suggested linkage disequilibrium between C2 deficiency, A10 and BW18. Awdeh et al. (1981) did C4 allotyping of 13 homozygous C2-deficient persons and found that 23 of 25 haplotypes were of the relatively rare type C4A*4B*2. ... Animal Model Although guinea pigs with C2 and C4 deficiency appeared healthy, Bottger et al. (1986) found that they had serologic characteristics of immune complex disease.
-
Cryoglobulinemia
Wikipedia
It has been proposed that these cases be termed an intermediate type II-III variant of cryoglobulinemic disease and that some of the type III cases associated with the expression of low levels of a one or more isotypes of circulating monoclonal immunoglobulin(s) are in transition to type II disease. [7] [10] Signs and symptoms [ edit ] The clinical features of cryoglobulinemic disease can reflect those due not only to the circulation of cryoglobulins but also to any underlying hematological premalignant or malignant disorder, infectious disease, or autoimmune syndrome. ... Mechanism [ edit ] Cryoglobulins [ edit ] Cryoglobulins consists of one or more of the following components: monoclonal or polyclonal IgM , IgG , IgA antibodies, monoclonal κ , or λ free light chain portions of these antibodies, and proteins of the blood complement system , particularly complement component 4 (C4). The particular components involved are a reflection of the disorders which are associated with, and considered to be the cause of, the cryoglobulinemic disease. ... Non-IgM monoclonal immunoglobulin-based cases of cryoglobulinemic disease are less commonly associated with other B-cell lymphocytic diseases viz., Non-Hodgkin lymphoma , Hodgkin lymphoma , B-cell chronic lymphocytic leukemia , and Castleman disease ; they occur rarely in non-B cell hematological disorders such as myelodysplastic syndromes and chronic myelogenous leukemia . [7] Among these purely monoclonal immunoglobulin causes of cryoglobulinemic disease, Waldenström macroglobulinemia and multiple myeloma together account for ≈40% of cases; their pre-malignant precursors account for ≈44% of cases; and the other cited hematological diseases account for ≈16% of cases. [2] [3] Mixtures of monoclonal or polyclonal IgM, IgG, and/or IgA along with blood complement proteins such as C4 are the cryoglobulins associated with cases of infectious diseases, particularly hepatitis C infection, HIV infection, and Hepatitis C and HIV coinfection , and, less commonly or rarely, with cases of other infectious diseases such as hepatitis B infection, hepatitis A infection, cytomegalovirus infection, Epstein–Barr virus infection , Lyme disease , syphilis , lepromatous leprosy , Q fever , poststreptococcal nephritis , subacute bacterial endocarditis , coccidioidomycosis , malaria , schistosomiasis , echinococcosis , toxoplasmosis , and Kala-azar . These mixed-protein cryoglobulins are also associated with autoimmune diseases , particularly Sjögren syndrome , less commonly systemic lupus erythematosus and rheumatoid arthritis , and rarely polyarteritis nodosa , systemic sclerosis , temporal arteritis , polymyositis , Henoch–Schönlein purpura , pemphigus vulgaris , sarcoidosis , inflammatory bowel diseases , and others. [7] In these mixed-protein depositions, the monoclonal or polyclonal IgM typically possesses rheumatoid factor activity and therefore binds to the Fc region of polyclonal IgG antibodies, activates the blood complement system, and complexes with complement components to form precipitates composed of IgM, IgG or IgG, and complement components, particularly complement component 4 (C4). [3] Diagnosis [ edit ] Cryoglobulinemia and cryoglobulinemic disease must be distinguished from cryofibrinogenemia or cryofibrinogenemic disease, conditions which involve the cold-induced intravascular deposition of circulating native fibrinogens. [13] [14] These molecules precipitate at lower temperatures (e.g., 4 °C). ... Other routine tests include measuring blood levels of rheumatoid factor activity, complement C4, other complement components, and hepatitic C antigen .TSLP, NOTCH4, MYD88, HLA-DRB1, CXCL10, TNF, GPT, IFNA13, IFNL3, IFNA1, IFNG, TLR2, NLRP3, CCR5, BTG3, TNFSF13B, IVNS1ABP, CXCL13, NR1D2, IL1R2, VIPR1, VCAM1, CRYGD, SMN2, IGHG3, SMN1, CCL2, PTPN11, PAEP, BCL2, HLA-A, MYC, LAG3, KIR3DL1, ITGAM, IL1RN, ADK

-
Hemolytic Uremic Syndrome, Atypical, Susceptibility To, 6
OMIM
Four patients had decreased serum C3 (120700), consistent with activation of the alternative complement pathway. C4 (120810) levels were normal. Molecular Genetics In 7 (4.6%) of 153 patients with aHUS, Delvaeye et al. (2009) identified 6 different heterozygous mutations in the THBD gene (see, e.g., 188040.0005-188040.0008).CFH, DGKE, CD46, C3, CFB, THBD, CFHR3, CFHR1, CFI, VTN, BAAT, ADAMTS13, TRIM25, C17orf67, CAPG, C5, CFHR5, VWF, C4BPA, C5AR1, IGAN1, PLG, C4BPB, CRP, GRHPR, CFHR4, C3AR1, TNF, GPR182, CABIN1, HPLH1, SMARCAL1, KRT20, SPZ1, PRSS55, CRISP2, THBS1, CRYGD, HP, ETFA, FHL1, MTOR, G6PD, GPI, CR1, CPB1, CD40LG, SPTA1, IL5, CD36, MUC1, MS4A1, NHS, PIGA, MASP1, NFE2L2
-
Meier-Gorlin Syndrome 7
OMIM
INHERITANCE - Autosomal recessive GROWTH Height - Short stature Weight - Low weight Other - Growth failure, progressive HEAD & NECK Head - Craniosynostosis - Microcephaly, progressive Ears - Microtia - Hearing loss Eyes - Thin eyebrows - Proptosis - Strabismus - Myopia Nose - Choanal atresia Mouth - Small mouth - High palate - Cleft palate CARDIOVASCULAR Heart - Atrial septal defect - Ventricular septal defect - Atrioventricular canal - Atrioventricular conduction block RESPIRATORY Lung - Pulmonary hypoplasia (in 1 patient) CHEST Breasts - Breast agenesis ABDOMEN Gastrointestinal - Anterior anus - Anal stenosis - Imperforate anus - Anorectal malformation - Duodenal stenosis GENITOURINARY External Genitalia (Male) - Hypospadias - Micropenis - Urethral stricture External Genitalia (Female) - Clitoromegaly Internal Genitalia (Male) - Undescended testes Ureters - Vesicoureteral reflux SKELETAL Skull - Microcephaly, progressive - Unicoronal or bicoronal craniosynostosis - Lambdoid or bilateral lambdoid craniosynostosis - Sagittal craniosynostosis - Large anterior fontanel - Copper-beaten appearance of skull Spine - Scoliosis (in 1 patient) - C1-C3 fusion (in 1 patient) - C4-C7 fusion (in 1 patient) - Thoracic vertebral segmentation defects (in 1 patient) Limbs - Patellar aplasia/hypoplasia - Bilateral radial head dislocation - Bowed legs (in 1 patient) - Joint laxity (in 1 patient) Hands - Digital clubbing (in 1 patient) - Syndactyly of second, third, and fourth fingers, mild (in 1 patient) - Preaxial polydactyly, bilateral (in 1 patient) Feet - Syndactyly of second and third toes NEUROLOGIC Central Nervous System - Developmental delay, mild to severe - Chiari I malformation (in 1 patient) MOLECULAR BASIS - Caused by mutation in the cell division cycle 45, S. cerevisiae, homolog-like gene (CDC45L, 603465.0001 ) ▲ CloseORC1, CDT1, GMNN, ORC6, CDC6, ORC4, CDC45, BMP5, DRD2, SLC6A3, CYP2D6, MAPK1, MYH9, TLR4, MTOR, RBP4, MCM5, PSEN1, COMT, PTH, MAPK3, SOD1, RELA, PRKCA, PRKACG, RGS2, ATXN2, SLC18A2, VEGFA, SYT1, TGFB1, TNF, PIK3CG, PDPN, POSTN, CHEK2, PLCB1, SIRT1, ORC3, DISC1, CLEC7A, GORASP1, WNK1, PLA2G4A, ACTG1, PIK3CA, HTR2A, ASPH, BRCA2, BSG, CD2, COL1A1, CRP, CCN2, DRD3, EPHB2, FANCD2, FASN, FOXM1, GNAS, GRIK3, IL6, SERPINE1, IL10, INSR, ITPR1, LEP, CYP4F3, LY6E, LYZ, MAOB, MCM2, ATXN3, NOS2, ACTB, APOE, PAEP, ACHE
-
Human Papillomavirus Type 18 Integration Site 2
OMIM
In 2 cervical carcinoma cell lines, HeLa and C4-I, HPV18 DNA was integrated in chromosome 8, 5-prime to the MYC gene (190080).





