Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Complement Component 4b Deficiency
OMIM
Of 26 patients with autoimmune chronic active hepatitis beginning in childhood, Vergani et al. (1985) found low C4 in 18 (69%) and low C3 serum levels in 5 (19%). ... Molecular Genetics Awdeh et al. (1981) analyzed C4 types in relatives of a C4-deficient proband and provided evidence that the deficiency results from homozygosity for a rare, double-null haplotype. The family contained persons with 1, 2, 3, or 4 expressed C4 genes, and the mean serum C4 levels roughly reflected the number of structural genes present. ... They confirmed the earlier findings of high frequencies of C4-null phenotypes and of HLA-B8,DR3 antigens. ... Although complete homozygous deficiency of complement C4 is one of the strongest genetic risk factors for SLE, partial C4 deficiency states do not independently predispose to the disease.
-
Complement Component 4a Deficiency
OMIM
Of 26 patients with autoimmune chronic active hepatitis beginning in childhood, Vergani et al. (1985) found low C4 in 18 (69%) and low C3 serum levels in 5 (19%). ... Molecular Genetics Awdeh et al. (1981) analyzed C4 types in relatives of a C4-deficient proband and provided evidence that the deficiency results from homozygosity for a rare, double-null haplotype. The family contained persons with 1, 2, 3, or 4 expressed C4 genes, and the mean serum C4 levels roughly reflected the number of structural genes present. ... They confirmed the earlier findings of high frequencies of C4-null phenotypes and of HLA-B8,DR3 antigens. ... Although complete homozygous deficiency of complement C4 is one of the strongest genetic risk factors for SLE, partial C4 deficiency states do not independently predispose to the disease.
-
Blood Group, Chido/rodgers System
OMIM
Description The blood groups Chido (Ch) and Rodgers (Rg) are epitopes on the C4 protein, and polymorphisms associated with these epitopes may lead to the formation of antibodies to the Ch or Rg antigens in transfused patients. Identification of anti-Ch or anti-Rg is based on antibody neutralization with plasma from Ch-positive or Rg-positive individuals and lack of reactivity with qualified Ch-negative or Rg-negative red blood cells. The C4 protein occurs in 2 forms, C4A and C4B, which are encoded by 2 closely linked genes. ... Nine antigens have been described for the Ch/Rg system: 6 of high prevalence for Ch, 2 of high prevalence for Rg, and 1 of low prevalence, WH. Eight phenotypes of the Ch/Rg system have been established, with 88.2% of individuals having the Ch(1,2,3) Chido phenotype and 95.0% of individuals having the Rg(1,2) Rodgers phenotype (review by Mougey (2010)). ... History The Chido blood group, which was discovered by Harris et al. (1967), is an antigenic characteristic of C4B. Chido has a low frequency of negatives (2%) and is tightly linked to HLA (Middleton and Crookston, 1972), closer to HLA-B (142830) than to HLA-A (142800). The Chido antigen resembles the HLA antigens in molecular structure. Like Chido, Rodgers has a low frequency of negatives (about 3%) and is closely linked to HLA (Giles et al., 1976).
-
Leukotriene C4 Synthase Deficiency
Wikipedia
Leukotriene C4 synthase deficiency Leukotriene C4 Leukotriene C4 synthase deficiency is an inborn error of metabolism . [1] [2] Deficiency of Leukotriene C4 synthase can lead to a reduction in Leukotriene C4 . References [ edit ] ^ Mayatepek E, Flock B (November 1998). "Leukotriene C4-synthesis deficiency: a new inborn error of metabolism linked to a fatal developmental syndrome". ... External links [ edit ] Classification D OMIM : 246530 MeSH : C565439 v t e Eicosanoid metabolism disorders Prostanoids PTGIS ( Essential hypertension ) TBSAX1 ( Ghosal hematodiaphyseal dysplasia ) Leukotrienes LTC4s ( Leukotriene C4 synthase deficiency ) GGT1 ( Glutathionuria ) Other/ungrouped HPGD ( Primary hypertrophic osteoathropathy ) This article about an endocrine, nutritional, or metabolic disease is a stub .

-
Complement Component 4, Partial Deficiency Of
OMIM
Clinical Features Muir et al. (1984) described a partial deficiency of C4 in a kindred ascertained through a 26-year-old woman with systemic lupus erythematosus. Six healthy members of the family also had partial deficiency of C4. The inheritance pattern was autosomal dominant with involved persons in 4 sibships of 2 generations (and by inference in a third earlier generation) and with male-to-male transmission. This form of C4 deficiency differs from that in previously reported families in the mode of inheritance, in the marked reduction of C4 levels (2-5% of normal in the proband; 2.4-24.1% of normal in healthy relatives), and in the lack of linkage to HLA, BF and the C4 structural loci. Wisnieski et al. (1987) found no evidence of hypercatabolism of C4 in metabolic turnover studies which appeared to be compatible with C4 hyposynthesis, even though C4 structural alleles were intact in affected members. In kindred members with decreased C4 levels, Wisnieski et al. (1994) found that after a 15-minute incubation, approximately 50% of serum C1 inhibitor did not complex with and inhibit C1r.
-
Terminal Complement Pathway Deficiency
Wikipedia
You can help by adding to it . ( August 2017 ) Diagnosis [ edit ] Suspect terminal complement pathway deficiency with patients who have more than one episode of Neisseria infection. Complement tests C4 ( C ) FB ( A ) C3 CH50 Conditions · ↓ ↓ ↓ PSG , C3 NeF AA ↓ · ↓ · HAE , C4D · · · ↓ TCPD ↓ · /↓ ↓ ↓ SLE ↑ ↑ ↑ ↑ inflammation Initial complement tests often include C3 and C4, but not C5 through C9. Instead, the CH50 result may play a role in diagnosis: if the CH50 level is low but C3 and C4 are normal, then analysis of the individual terminal components may be warranted.
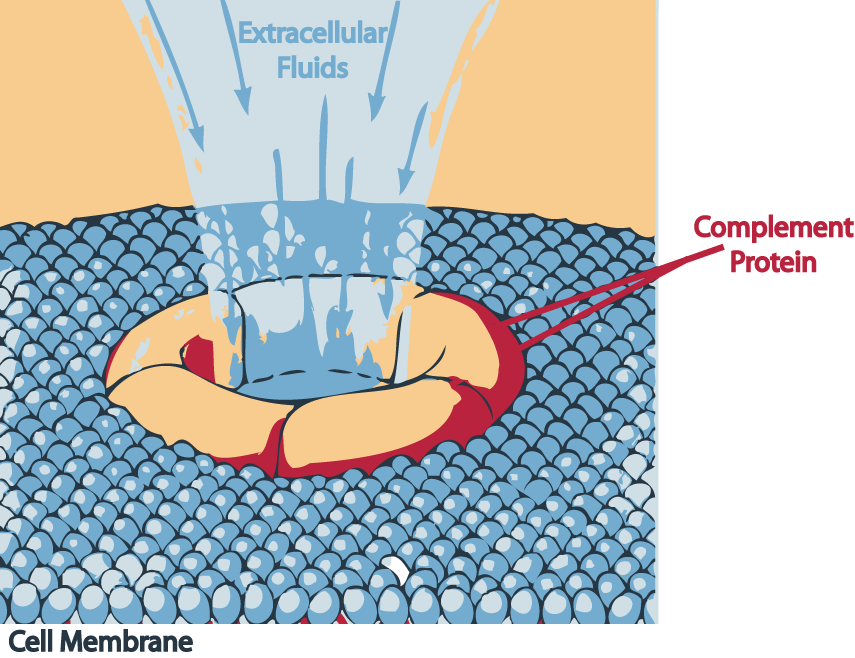
-
Hypocomplementemic Urticarial Vasculitis
Orphanet
Hypocomplementemic urticarial vasculitis (HUV) is an immune complex-mediated small vessel vasculitis characterized by urticaria and hypocomplementemia (low C1q with or without low C3 and C4), and usually associated with circulating anti-C1q autoantibodies. ... For example, patients with cutaneous disease and arthralgias but no major organ involvement may be managed with low-dose prednisone, hydroxychloroquine, or dapsone; whereas patients with major organ involvement, such as glomerulonephritis, may require high doses of corticosteroids and cytotoxic agents similar to the treatment for active SLE. Response to treatment is usually accompanied by a decrease in circulating anti-C1q titer and normalization of C3 and C4 levels, although C1q often remains low.
-
Acquired Angioedema With C1inh Deficiency
Orphanet
Clinical manifestation includes nonpitting edema of the skin predominantly involving the face, but also the limbs or genitals, as well as abdominal pain due to involvement of the gastrointestinal mucosa, and severe edema of the upper airway and oral mucosa. Laboratory examination shows low C1-INH activity and low C3, C4, and C1q levels.
-
Complement Deficiency
Wikipedia
MBL deficiency can be inherited by either manner. [2] Inherited [ edit ] Properdin deficiency is an X-linked [10] disorder that also causes susceptibility to Neisseria infections. [2] C1-inhibitor deficiency or hereditary angioedema will have low C4 with normal C1 levels. [11] Acquired [ edit ] Acquired hypocomplementemia may occur in the setting of bone infections (osteomyelitis) , infection of the lining of the heart (endocarditis) , and cryoglobulinemia . Systemic lupus erythematosus is associated with low C3 and C4 . [12] Membranoproliferative glomerulonephritis usually has low C3. [13] Mechanism [ edit ] Model of common structural genes and their possible contribution to the development of schizophrenia (as defined in the Sekar et al . article) The mechanism of complement deficiency consists of: C2 : In regard to C2 deficiency, about 5 different mutations in the C2 gene are responsible. ... This rare condition mutates or prevents C3 protein from forming, lowering the immune system's ability to protect. [15] C4 : C4 deficiency is highly associated with systemic lupus erythematosus . [3] Aβ42 , a protein involved in Alzheimer's disease , can cause activation of C4 (even in plasma deficient of C1q ). [16] At least one study indicates that the genetic variation of C4 plays a role in schizophrenia . [17] Diagnosis [ edit ] Complement tests C4 ( C ) FB ( A ) C3 CH50 Conditions · ↓ ↓ ↓ PSG , C3 NeF AA ↓ · ↓ · HAE , C4D · · · ↓ TCPD ↓ · /↓ ↓ ↓ SLE ↑ ↑ ↑ ↑ inflammation The diagnostic tests used to diagnose a complement deficiency include: [3] CH50 measurement Immunochemical methods/test C3 deficiency screening Mannose -binding lectin (lab study) Plasma levels/regulatory proteins (lab study) Types [ edit ] Disorders of the proteins that act to inhibit the complement system (such as C1-inhibitor ) can lead to an overactive response, causing conditions such as hereditary angioedema . [18] Disorders of the proteins that act to activate the complement system (such as C3 ) can lead to an underactive response, causing greater susceptibility to infections. [19] Treatment [ edit ] In terms of management for complement deficiency, immunosuppressive therapy should be used depending on the disease presented.C7, CFD, MASP2, C4A, C5, C6, C2, SERPING1, C9, CFI, CFH, CFHR1, CFHR2, CFB, CFP, THBD, FCN3, CFHR4, CFHR3, CD46, CFHR5, CD55, CD59, C1QA, C1QB, C1QC, C1R, C1S, C3, C4B, C8A, C8B, C8G, SLC7A7, DNASE1L3, MTA2, IL1RN, IL1B, IL1A, TRIM21, CALR, ACE

-
Acquired Angioedema Type 1
Orphanet
It is often associated with lymphoproliferative or autoimmune diseases which produce immune factors that destroy C1-INH leading to low levels of C1-INH, C1q complement and C4 complement.
-
Isobutyryl-Coa Dehydrogenase Deficiency
OMIM
Clinical Features The first patient with isobutyryl-CoA dehydrogenase deficiency was described by Roe et al. (1998) and presented at age 12 months with dilated cardiomyopathy, anemia, and carnitine deficiency. An elevated C4-acylcarnitine was noted in a plasma acylcarnitine profile, but a subsequent urine organic acid analysis was normal. ... All were identified by newborn screening when isolated elevation of C4-acylcarnitine prompted further diagnostic investigations. ... Oglesbee et al. (2007) reported an additional 13 patients with IBD deficiency who were identified by newborn screening due to an elevation of C4-acylcarnitine in dried blood spots. ... Diagnosis Oglesbee et al. (2007) described an algorithm for the diagnosis of IBD deficiency identified by elevated C4-acylcarnitine on newborn screening.

-
3-Hydroxyisobutyryl-Coa Hydrolase Deficiency
OMIM
This was viewed as an example of an inborn error of metabolism with effects due to intracellular accumulation of a small amount of a highly toxic intermediate metabolite rather than the accumulation in the body fluids of high levels of metabolites of relatively low toxicity. Recessive inheritance seemed quite certain because both parents had intermediate levels of enzyme activity. ... Laboratory studies showed increased lactate; 1 patient had increased hydroxy-C4-carnitine. Skeletal muscle biopsy showed decreased activity of mitochondrial respiratory enzyme complexes II, II+III, and IV as well as decreased activity of the pyruvate dehydrogenase complex (PDC) in 1 patient; these values were normal in the other patient. ... The results of laboratory investigations and muscle biopsy were inconsistent at first, but eventually showed increased blood lactate, increased blood hydroxy-C4-carnitine, and depletion of mtDNA and decreases in multiple mitochondrial respiratory enzyme activities in skeletal muscle. ... Stiles et al. (2015) demonstrated elevated hydroxy-C4-carnitine in newborn screening cards for these 2 sibs. ... Molecular Genetics Using immunoblot analysis, Loupatty et al. (2007) demonstrated absence of the HIBCH protein in the patient reported by Brown et al. (1982) and an apparently low expression of the HIBCH protein in their own patient.

-
Lipodystrophy, Partial, Acquired, With Low Complement Component C3, With Or Without Glomerulonephritis
OMIM
See 608709 for a subtype of APLD not associated with low complement C3 or renal disease. Clinical Features Alper et al. (1973) reported a 30-year-old woman with a lifelong history of recurrent bacterial infections who was found to have low serum C3 due to presence in the serum of an activator of C3, which the authors termed hypercatabolism. ... Laboratory studies showed that 17 had low serum C3, accompanied in 14 by C3 nephritic factor, suggesting activation of the alternative complement pathway. ... Kidney biopsy showed membranoproliferative glomerulonephritis, and laboratory values showed decreased serum C3 and C4, as well as proteinuria, hematuria, and leukocyturia. ... The median age at onset of lipodystrophy was 7 years, and the female to male ratio was 4:1. Approximately 83% of patients had low serum C3 with the presence of the C3 nephritic factor autoantibody. ... Serum C3 was approximately one-third of normal values, while C4 was normal. Reichel et al. (1976) noted that the patient had a susceptibility to infection, with a history of pneumonia, meningitis, encephalitis, and viral hepatitis.
-
Short-Chain Acyl-Coa Dehydrogenase Deficiency
GeneReviews
Positive Newborn Screening (NBS) Result NBS for SCADD is primarily based on acylcarnitine analysis by tandem mass spectrometry to detect elevated blood C4 (butyrylcarnitine). Note: (1) Normal ranges for isolated C4 vary from state to state, necessitating confirmatory testing consistent with the American College of Medical Genetics (ACMG) ACT Sheet. (2) Isobutyryl-CoA dehydrogenase deficiency (IBDD) that leads to elevation of isobutyrylcarnitine, a C4 species also detectable by NBS, must be distinguished from SCADD by additional laboratory testing. C4 values above the cutoff reported by the screening laboratory are considered positive and require additional biochemical testing and in most cases molecular genetic testing to establish the diagnosis (see Establishing the Diagnosis). ... In addition, three parents and six sibs were found to have ACADS genotypes identical to the proband; eight of the nine had increased levels of C4 and/or EMA, and one of the six sibs had transient feeding difficulties in the first year. ... IBDD, also detectable by NBS, leads to elevation of isobutyrylcarnitine, a C4 species indistinguishable from butyrylcarntine without additional separation techniques. ... Ethylmalonic encephalopathy (OMIM 602473) presents with ethylmalonic acid in urine at much higher levels than in SCADD. C4 may be higher, but as in GAII, other metabolites may be elevated.ACADS, SERPINA5, SMAD3, NME1, MIR34A, SOST, ACAD8, VEGFA, TLN1, TGFB1, PLXNA2, MPO, ACADSB, LAD1, ITGB2, F3, DLD, AUH, ANXA1, AK4, LINC01672
-
Complement Factor H Deficiency
OMIM
The factor H deficiency was defined by undetectable complement hemolytic activity by the classic (CH50) and alternate (AP50) pathways, and low levels of C3 and factor B (138470). ... Lopez-Larrea et al. (1987) studied a family in which 3 female sibs had undetectable levels of factor H and C3 nephritic factor, low levels of factor B, C3, and C5 (see 120500), and normal levels of C4-binding protein (120830), factor I (217030), and classic pathway factors. C4 (see 120810) levels were low in 1 patient. ... Among 21 relatives of the proband encompassing 3 generations, 10 had low factor H levels, including 2 children of the proband, indicating heterozygosity. ... Decreased levels of serum C3 and factor B but normal levels of serum C4 and factor I were found; factor H was undetectable by radial immunodiffusion analysis.
-
Klippel-Feil Syndrome 1, Autosomal Dominant
OMIM
The clinical triad consists of short neck, low posterior hairline, and limited neck movement, although less than 50% of patients demonstrate all 3 clinical features (Tracy et al., 2004). ... Vertebral fusion was confined to the cervical spine in all affected members at C2-3; C4-5 and C6-7 fusions were less consistently present. ... In total, 11 family members had radiographically confirmed C2-3 fusion, 70% of whom had C2-3 and C4-5 skipped fusion, and 20% of whom had C2-3, C4-5, and C6-7 skipped fusion. ... Clarke et al. (1998) reported a family in which the proband had vertebral fusion at C4-5 and her father had fusion at C5-6. ... The affected twin had short neck, limited neck movement, and low posterior hairline. CT scan showed C1-4 vertebral fusion.

-
Cryoglobulinemic Vasculitis
Wikipedia
Due to deposition of complement (in particular, C4), low levels of circulating complement factors may be seen.CR2, IFNA13, IFNA1, CD27, IFNL3, CDR3, IGHG3, CXCR3, TNFSF13B, ITGAX, CXCL10, KRT20, MS4A1, BCL2, MPIG6B, FCRL4, BHLHE40, TNFRSF25, CXCL13, MIR21, IGHV4-59, IGHV1-69, CENPX, RBM45, DELEC1, GDE1, RTEL1, MIR155, TSLP, WDR11, VEGFA, SERPINA13P, PAEP, VDR, IFNG, CD40LG, CD44, CLU, CRYGD, FBL, EXT1, FCGR3B, FN1, HLA-DRB1, IGH, TNF, IL1B, IL6, IL13, LAG3, MIP, NOTCH4, BCL6, ABCB1, TLR2, MIR26B

-
Acquired Generalized Lipodystrophy
Orphanet
Biologically, hyperinsulinemia and insulin-resistant diabetes are observed, often associated with severe hypertriglyceridemia with low plasma levels of leptin and adiponectin. ... A recent publication showed activation of the classical complement pathway (low C4). This is in contrast to acquired partial lipodystrophy (see this term) which affects the upper half of the body and is characterized by an activation of the alternative complement pathway (low C3).
- Acquired Angioedema Orphanet
-
Non-Histaminic Angioedema
Orphanet
Diagnostic methods Diagnosis is based on clinical findings, measurement of C4 concentrations and on quantitative and functional analysis of C1-INH. C1q levels are low in patients with AAE but are normal in patients with HAE.XPNPEP2, ALB, HLA-DPB1, PLAT, HLA-DRB1, HLA-DQB1, F12, GRK6, SERPING1, DNASE1L3, CPN1, PLG, COL4A1, SPINK5, ABI3BP, SYTL2, IFT43, DPP4