Bilateral Frontoparietal Polymicrogyria
Bilateral frontoparietal polymicrogyria is a genetic disorder with autosomal recessive inheritance that causes a cortical malformation. Our brain has folds in the cortex to increase surface area called gyri and patients with polymicrogyri have an increase number of folds and smaller folds than usual. Polymicrogyria is defined as a cerebral malformation of cortical development in which the normal gyral pattern of the surface of the brain is replaced by an excessive number of small, fused gyri separated by shallow sulci and abnormal cortical lamination. From ongoing research, mutation in GPR56, a member of the adhesion G protein-coupled receptor (GPCR) family, results in BFPP. These mutations are located in different regions of the protein without any evidence of a relationship between the position of the mutation and phenotypic severity. It is also found that GPR56 plays a role in cortical pattering.
Presentation

- Symptoms: Developmental delay, Psychomotor delay, Mental retardation - moderate to severe, Exaggerated reflexes and Seizures (epilepsy)
Associated conditions
BFPP is a cobblestone-like cortical malformation of the brain. Disruptions of cerebral cortical development due to abnormal neuronal migration and positioning usually lead to cortical disorders, which includes cobblestone lissencephaly. Cobblestone lissencephaly is typically seen in three different human congenital muscular dystrophy syndromes: Fukuyama congenital muscular dystrophy, Walker-Warburg syndrome, and muscle-eye-brain disease. In cobblestone lissencephaly, the brain surface actually has a bumpy contour caused by the presence of collections of misplaced neurons and glial cells that have migrated beyond the normal surface boundaries of the brain. Sometimes regions populated by these misplaced cells have caused a radiologic misdiagnosis of polymicrogyria. However, the presence of other abnormalities in these cobblestone lissencephaly syndromes, including ocular anomalies, congenital muscular dystrophy, ventriculomegaly, and cerebellar dysplasia, usually distinguishes these disorders from polymicrogyria. There are no anatomopathologic studies that have characterized the pattern of cortical laminar alterations in patients with GPR56 gene mutations, but it has been suggested that the imaging characteristics of BFPP, including myelination defects and cerebellar cortical dysplasia, are reminiscent of those of the so-called cobblestone malformations (muscle-eye-brain disease and Fukuyama congenital muscular dystrophy) that are also associated with N-glycosylation defects in the developing brain.
Lissencephaly ("smooth brain") is the extreme form of pachygyria. In lissencephaly, few or no sulci are seen on the cortical surface, resulting in a broad, smooth appearance to the entire brain. Lissencephaly can be radiologically confused with polymicrogyria, particularly with low-resolution imaging, but the smoothness and lack of irregularity in the gray-white junction, along with markedly increased cortical thickness, distinguishes lissencephaly.
GPR56 mutation also can cause a severe encelphalopathy which is associated with electro clinical features of the Lennox-Gastaut syndrome. Lennox-Gastaut syndrome can be cryptogenic or symptomatic, but the symptomatic forms have been associated with multiple etiologies and abnormal cortical development. BFPP caused by GPR56 mutations is a manifestation of a malformation of cortical development that causes Lennox-Gastaut Syndrome.
Polymicrogyria is often confused with pachygyria; therefore, it needs to be distinguished from pachygyria, a distinct brain malformation in which the surface folds are excessively broad and sparse. Pachygyria and polymicrogyria may look similar on low-resolution neuroimaging such as CT because the cortical thickness can appear to be increased and the gyri can appear to be broad and smooth in both conditions. This is why higher resolution neuroimaging, such as an MRI, is necessary for proper diagnosis.
Genetics
The GPR56 is grouped in the B family of GPCRs. This GPCR group have long N termini characterized by an extracellular “cysteine box” and hydrophilic, potentially mucin-rich. The cysteine box contains four conserved cysteines and two tryptophans arranged in a specific fashion (C-x2-W-x6-16-W-x4-C-x10-22-C-x-C) just before the first transmembrane domain and serves as a cleavage site in some members of this group of G protein–coupled receptors. Although, the molecular and cellular mechanisms of how GPR56 regulates brain development remain largely unknown. These types of receptors play an essential role in biological processes including embryonic development, central nervous system (CNS), immune system, and tumorigenesis.

Mode of inheritance
Parents of a proband
- The parents of an affected individual are obligate heterozygotes and therefore carry one mutant allele.
- Heterozygotes (carriers) are asymptomatic.
Sibs of a proband
- At conception, each sibling of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Once an at-risk sibling is known to be unaffected, the risk of his/her being a carrier is 2/3.
- Heterozygotes (carriers) are asymptomatic.
Offspring of a proband
- Offspring of a proband are obligate heterozygotes and will therefore carry one mutant allele.
- In populations with a high rate of consanguinity, the offspring of a person with GPR56-related BFPP and a reproductive partner who is a carrier of GPR56-related BFPP have a 50% chance of inheriting two GPR56 disease-causing alleles and having BFPP and a 50% chance of being carriers.
Other family members of a proband.
- Each sibling of the proband's parents is at a 50% risk of being a carrier
Diagnosis
Diagnostic criteria for a BFPP patient entails a heterozygous genotype for a deletion of chromosome 16q12.1-q21 region, including GPR56 gene. To date the only gene known to be associated with polymicrogyria is GPR56. Testing for GPR56-related bilateral frontoparietal polymicrogyria is available clinically. Mutations in GPR56 hinders Collagen III, its specific ligand, to bind in a developing brain. To date, a total of fourteen BFPP-associated mutations have been identified, including one deletion, two splicing, and eleven missense mutations. Two mutations in the GPCR proteolytic site (GPS) domain, C346S and W349S, cause a brain malformation through trapping the mutated proteins in the endoplasmic reticulum.
GPR56 are a part of the B class of the GPCR family, the largest cell surface gene family in the human genome. Within this family there are different types of bio-active molecules that transduce their signal to the intracellular compartment via interaction with this type of receptor. Children often present with developmental delay, spasticity, or seizures; they are also often microcephalic. Some patients with polymicrogyria go undiagnosed until they produce children with the disorder who have more severe manifestations. Retrospectively, these patients will often report some difficulty in their medical or educational history. BFPP patients demonstrate mental retardation, language impairment, motor developmental delay, and seizure disorders such as epilepsy. The association of epilepsy is in approximately 50% to 85% of affected BFPP patients.
The clinical manifestations of polymicrogyria are stable neurologic deficits:
In the mildest form, polymicrogyria is unilateral with only one small region of the brain involved; neurologic problems may not be evident.
In more severe forms, focal motor, sensory, visual, or cognitive problems may be present, depending on the location of the brain region affected.
In the most severe forms, polymicrogyria is bilateral and generalized, resulting in severe intellectual disability, cerebral palsy, and refractory epilepsy.
Individuals with the milder forms of polymicrogyria survive into adulthood, while those with the most severe forms, such as BFPP, may die at a young age as a result of such complications as seizures or pneumonia. The prevalence of isolated polymicrogyria is unknown. Researchers believe that it may be relatively common overall, although BFPP is probably rare.
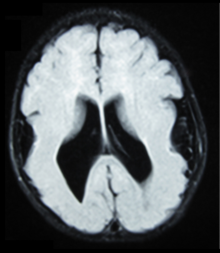
- Radiological findings (MRI) demonstrated symmetric generalized polymicrogyria with decreasing anterior-posterior gradient, most prominent in frontoparietal cortex.
- Numerous gyrus on the cortex
- Small gyri and sulci
- Thin cortex
Methods/tests

There are different tests or methods used to determine GPR56 expression or visuals of the brain to analyze the specific sections that are affected. These tests for example, using animals such as mice, RNAi, Behavioral assay, Electron microscopy, CT scan, or MRI demonstrate different results that concludes an affected BFPP patient. MRI's reveal either irregularity to the cortical surface suggestive of multiple small folds or an irregular, scalloped appearance of the gray matter-white matter junction.
Neuroimaging The diagnosis of polymicrogyria is typically made by magnetic resonance imaging (MRI) since computed tomography (CT) and other imaging methods generally do not have high enough resolution or adequate contrast to identify the small folds that define the condition. The cerebral cortex often appears abnormally thick as well because the multiple small gyri are fused, infolded, and superimposed in appearance.
Neuropathology Gross neuropathologic examination reveals a pattern of complex convolutions to the cerebral cortex, with miniature gyri fused and superimposed together, often resulting in an irregular brain surface. The cortical ribbon can appear excessively thick as a result of the infolding and fusion of multiple small gyri.
Microscopic examination demonstrates that the cerebral cortex is in fact abnormally thin and has abnormal lamination; typically the cortex is unlayered or has four layers, in contrast to the normal six layers. The most superficial layers between adjacent small gyri appear fused, with the pia (layer of the meninges) bridging across multiple gyri. Prenatal diagnosis for BFPP is also available for pregnancies at risk if the GPR56 mutations have been identified in an affected family member.
Treatment
Treatment plans will vary depending on the severity of the condition and its evidences in each patient. Areas that will probably need to be evaluated and assessed include speech, vision, hearing and EEG. Treatment measures may include physical therapy, occupational therapy, Speech therapy, anti-seizure drugs and orthotic devices. Surgery may be needed to assuage spastic motor problems. Various supportive measures such as joint contractures that could prevent complications. Genetic counseling may also be recommended
Prognosis
Once the diagnosis of polymicrogyria has been established in an individual, the following approach can be used for discussion of prognosis:
A pregnancy history should be sought, with particular regard to infections, trauma, multiple gestations, and other documented problems. Screening for the common congenital infections associated with polymicrogyria with standard TORCH testing may be appropriate. Other specific tests targeting individual neurometabolic disorders can be obtained if clinically suggested.
The following may help in determining a genetic etiology:
Family history
It is important to ask for the presence of neurologic problems in family members, including seizures, cognitive delay, motor impairment, pseudobulbar signs, and focal weakness because many affected family members, particularly those who are older, may not have had MRI performed, even if these problems came to medical attention. In addition, although most individuals with polymicrogyria do present with neurologic difficulties in infancy, childhood, or adulthood, those with mild forms may have no obvious deficit or only minor manifestations, such as a simple lisp or isolated learning disability. Therefore, if a familial polymicrogyria syndrome is suspected, it may be reasonable to perform MRI on relatives who are asymptomatic or have what appear to be minor findings. The presence of consanguinity in a child's parents may suggest an autosomal recessive familial polymicrogyria syndrome.
Physical examination
A general physical examination of the proband may identify associated craniofacial, musculoskeletal, or visceral malformations that could indicate a particular syndrome. Neurologic examination should assess cognitive and mental abilities, cranial nerve function, motor function, deep tendon reflexes, sensory function, coordination, and gait (if appropriate).
Genetic testing
See also
- Epilepsy Phenome/Genome Project