Kabuki Syndrome

Kabuki syndrome (also previously known as Kabuki-makeup syndrome (KMS) or Niikawa–Kuroki syndrome) is a congenital disorder of genetic origin. It affects multiple parts of the body, with varying symptoms and severity, although the most common is the characteristic facial appearance.
It is quite rare, affecting roughly one in 32,000 births. It was first identified and described in 1981 by two Japanese groups, led by scientists Norio Niikawa and Yoshikazu Kuroki. It is named Kabuki syndrome because of the facial resemblance of affected individuals to stage makeup used in kabuki, a Japanese traditional theatrical form.
Signs and symptoms
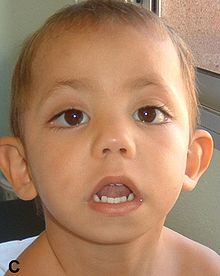
Specific symptoms for Kabuki syndrome vary, with large differences between affected individuals. Most people with Kabuki syndrome have distinctive facial features that include arched eyebrows, long eyelashes, elongated eyelids with lower lids that turn out, prominent ears, a flat tip of the nose and a downward slant to the mouth.
 a5/Child_with_Kabuki_Syndrome_08.jpg/220px-Child_with_Kabuki_Syndrome_08.jpg" decoding="async" width="220" height="147" class="thumbimage" srcset="//upload.wikimedia.org/wikipedia/commons/thumb/a/a5/Child_with_Kabuki_Syndrome_08.jpg/330px-Child_with_Kabuki_Syndrome_08.jpg 1.5x, //upload.wikimedia.org/wikipedia/commons/thumb/a/a5/Child_with_Kabuki_Syndrome_08.jpg/440px-Child_with_Kabuki_Syndrome_08.jpg 2x" data-file-width="960" data-file-height="640">
a5/Child_with_Kabuki_Syndrome_08.jpg/220px-Child_with_Kabuki_Syndrome_08.jpg" decoding="async" width="220" height="147" class="thumbimage" srcset="//upload.wikimedia.org/wikipedia/commons/thumb/a/a5/Child_with_Kabuki_Syndrome_08.jpg/330px-Child_with_Kabuki_Syndrome_08.jpg 1.5x, //upload.wikimedia.org/wikipedia/commons/thumb/a/a5/Child_with_Kabuki_Syndrome_08.jpg/440px-Child_with_Kabuki_Syndrome_08.jpg 2x" data-file-width="960" data-file-height="640">  b2/Child_with_Kabuki_Syndrome_03.jpg/220px-Child_with_Kabuki_Syndrome_03.jpg" decoding="async" width="220" height="293" class="thumbimage" srcset="//upload.wikimedia.org/wikipedia/commons/thumb/b/b2/Child_with_Kabuki_Syndrome_03.jpg/330px-Child_with_Kabuki_Syndrome_03.jpg 1.5x, //upload.wikimedia.org/wikipedia/commons/thumb/b/b2/Child_with_Kabuki_Syndrome_03.jpg/440px-Child_with_Kabuki_Syndrome_03.jpg 2x" data-file-width="720" data-file-height="960">
b2/Child_with_Kabuki_Syndrome_03.jpg/220px-Child_with_Kabuki_Syndrome_03.jpg" decoding="async" width="220" height="293" class="thumbimage" srcset="//upload.wikimedia.org/wikipedia/commons/thumb/b/b2/Child_with_Kabuki_Syndrome_03.jpg/330px-Child_with_Kabuki_Syndrome_03.jpg 1.5x, //upload.wikimedia.org/wikipedia/commons/thumb/b/b2/Child_with_Kabuki_Syndrome_03.jpg/440px-Child_with_Kabuki_Syndrome_03.jpg 2x" data-file-width="720" data-file-height="960"> Other common symptoms are skeletal abnormalities, short stature, heart defects, feeding difficulties and a failure to thrive, vision and hearing difficulties, weak muscle tone (hypotonia), small head size (microcephaly), and frequent infections.
Mild to moderate intellectual disability and mild to severe developmental delay are often associated with Kabuki syndrome. Infants and young children often experience difficulties relating to hypotonia, feeding issues/failure to thrive, infections, surgical repair of heart and palate defects and developmental delays.
Young children with Kabuki syndrome benefit from early intervention services. School age children tend to have fewer medical issues requiring hospitalization, though frequent infections, hearing loss and feeding issues occur. In addition, intellectual impairment, difficulty with visuospatial tasks and maintaining attention usually require an IEP (individualized education plan) if the child attends public school. Older children and adults report difficulties with anxiety. Endocrine abnormalities and immune system abnormalities such as ITP (idiopathic thrombocytopenia) and CVID (common variable immune deficiency) are medical issues that tend to present in older children, adolescents and adults.
Causes
Kabuki syndrome is one of the Mendelian disorders of epigenetic machinery. Most cases of Kabuki syndrome occur de novo, that is, the parents are unaffected and the gene was mutated early in embryological development. It is classified as a Mendelian disorder because individuals who have a de novo mutation in a specific gene pass the mutation to offspring according to the laws of Mendelian inheritance. There are two known genes that cause Kabuki syndrome: KMT2D and KDM6A. However, about 30% of cases have no identifiable causative mutation.
It is estimated that between 55-80% of cases of Kabuki syndrome are caused by mutations in the KMT2D gene, formerly known as the MLL2 gene. This gene is located on chromosome 12. A mutation in the KMT2D gene results in a non functional lysine (K)-specific methyltransferase 2D enzyme and demonstrates an autosomal dominant pattern of inheritance. Another 2-6% of cases are related to mutations in the KDM6A gene, located on the X chromosome. This mutation produces a nonfunctional lysine (K)-specific demethylase 6A enzyme and demonstrates an X-linked dominant pattern of inheritance.
Pathophysiology
The KMT2D and KDM6A genes belong to a family of genes called chromatin-modifying enzymes. Specifically, these genes code for a histone methyltransferase (KMT2D) and a histone demethylase (KDM6A), and play a part in the regulation of gene expression. Under normal circumstances, these enzymes transfer methyl groups on and off histones to regulate genes via epigenetic pathways. When the genes that encode these enzymes are mutated, epigenetic activation of certain developmental genes is impaired and developmental abnormalities occur, leading to the characteristics of Kabuki syndrome patients. The specific developmental genes that are affected by the impaired epigenetic mechanisms in Kabuki syndrome are not yet fully known.
There are hundreds of different mutations that have been identified in Kabuki syndrome patients. Most of these mutations are in the KMT2D gene and involve a change in amino acid sequence that creates a shortened and nonfunctional chromatin-modifying enzyme.
Diagnosis
A consensus on clinical diagnostic criteria for Kabuki syndrome (KS) was defined in December 2018 by an international group of experts. The authors propose that a definitive diagnosis can be made in an individual of any age with a history of infantile hypotonia, developmental delay and/or intellectual disability, and one or both of the following major criteria: (1) a pathogenic or likely pathogenic variant in KMT2D or KDM6A; and (2) typical dysmorphic features (defined below) at some point of life. Typical dysmorphic features include long palpebral fissures with eversion of the lateral third of the lower eyelid and two or more of the following: (1) arched and broad eyebrows with the lateral third displaying notching or sparseness; (2) short columella with depressed nasal tip; (3) large, prominent or cupped ears; and (4) persistent fingertip pads. Further criteria for a probable and possible diagnosis, including a table of suggestive clinical features, were included in the publication.
The original description of Kabuki syndrome by Niikawa et al. defined five cardinal manifestations, although some of these “cardinal manifestations” may or may not be present in a patient with Kabuki syndrome.
- Typical facial features: Elongated palpebral fissures with eversion of the lateral third of the lower eyelid; arched and broad eyebrows with the lateral third displaying sparseness or notching; short columella with depressed nasal tip; large, prominent, or cupped ears
- Skeletal anomalies: Spinal column abnormalities, including sagittal cleft vertebrae, butterfly vertebrae, narrow intervertebral disc space, and/or scoliosis, Brachydactyly V Brachymesophalangy Clinodactyly of fifth digits
- Dermatoglyphic abnormalities: persistence of fetal fingertip pads
- Mild to moderate intellectual disability
- Postnatal growth deficiency
Kabuki syndrome is diagnosed clinically (through identifying symptoms, physical exams, and lab results), most commonly by a geneticist. Alternatively, it may be discovered using genetic testing (whole exome or whole genome sequencing).
Diagnosis can be difficult given the large spectrum of disease. The fact that some patients do not carry one of the two known mutations or can carry multiple mutations complicates the diagnosis further.
Screening
Due to its rarity, Kabuki syndrome is not screened for in routine prenatal testing including blood tests, chorionic villus sampling (CVS), or amniocentesis. Although not routine for the general population, if Kabuki syndrome is a specific concern (i.e. expectant mother who has been diagnosed with Kabuki syndrome or sibling with KS), it is possible to test for one of the specific mutations. This prenatal testing does require a CVS or amniocentesis. However Kabuki syndrome is usually not inherited and therefore most cases do not have a positive family history. Kabuki syndrome can have positive screening tests, such as cystic hygroma seen on nuchal translucency ultrasound screening, although these findings are non-specific and have a wide differential diagnosis.
Management
Newly diagnosed patients with Kabuki syndrome will often undergo tests that are aimed at detecting common abnormalities associated with the syndrome. They include an echocardiogram (ultrasound of the heart) for detection of structural heart defects, kidney ultrasound for detection of structural renal abnormalities, immunoglobulin levels, pneumococcal titers and a hearing screening test.Further evaluation and testing by specialists may be indicated in addition to cardiology, nephrology, allergy/immunology, audiology-mentioned above. This may include orthopedics (such as hip dysplasia), pulmonary (sleep study to rule out obstructive sleep apnea due to hypotonia), ophthalmology evaluation (vision screen), ENT evaluation (hearing evaluation), Neurology evaluation (i.e. if seizures present), Hematology evaluation (if bleeding disorder), GI evaluation (if gi abnormalities), or others as needed.
There is no specific treatment for Kabuki syndrome. Treatment plans are customized to address the symptoms the individual is experiencing. For example, someone experiencing seizures will be treated with the standard anti-epilepsy therapies. Additionally, patients with Kabuki syndrome are routinely evaluated and monitored to address problems that may develop, such as vision or hearing problems, or cognitive difficulties. If congenital heart disease is present, prophylactic antibiotics may be recommended before any procedures such as dental work that might cause infection.
Prognosis
The life expectancy of individuals with Kabuki syndrome is not shortened by the syndrome in most cases. Some patients have coexisting conditions which may shorten life expectancy, such as hypoplastic left heart syndrome or kidney dysfunction. It is important that patients with cardiac, renal, or immunologic issues are identified and well-managed.
Epidemiology
Kabuki syndrome occurs about once in every 32,000 births. The disease appears to affect all population groups equally, with no differences based on sex, race, or environment.
Research
Research on Kabuki syndrome is extremely limited due to its low incidence. Despite this, several groups around the world are studying Kabuki syndrome. In the United States, these include the Epigenetics and Chromatin Clinic at Johns Hopkins University (led by Dr. Hans Bjornsson), The Roya Kabuki Program at Boston Children's Hospital, Dr. Mark Hannibal at the University of Michigan, groups at University of Colorado, University of Utah, University of South Florida and others.
History
In 1969, Norio Niikawa MD, a geneticist in Japan was treating a child patient presenting with unique facial characteristics and various health problems. Never having seen this constellation of symptoms before, Dr Niikawa wondered if he was faced with an undiagnosed condition, a disorder with a genetic basis. Over the next several years, this physician treated several other patients with the same symptoms in his outpatient genetics clinic, furthering support for a disorder never before diagnosed.
In 1979, Dr Niikawa presented his findings and hypothesis at the first Japan Dysmorphology Conference. A fellow physician at this conference, Yoshikazu Kuroki, recognised the symptoms, and realised that he had also seen several paediatric patients with this presentation; he presented two of his own cases at the second annual conference the following year. In 1981, the two doctors separately submitted articles on this new diagnosis to the Journal of Pediatrics.
Dr Niikawa coined the term ‘Kabuki syndrome’ (also known as Kabuki make-up syndrome or Niikawa–Kuroki syndrome) as a reference to traditional Japanese theatre which he respected greatly. Many of the children presenting with this diagnosis had unusual, elongated lower eyelids, and this feature was reminiscent of the theatrical make-up worn by actors in Kabuki theatre.
As reported by Dr. Niikawa "The name, “Kabuki make-up”, of the syndrome was given by myself, because the facial appearance of patients, especially eversion of their lower eyelids, is reminiscent of the makeup of actors in Kabuki, the traditional form of Japanese theater. Kabuki was founded early in the 17th century in Japan and over the next 300 years developed into a sophisticated form of theater. Kabuki actors usually apply traditional makeup to strengthen their eyes, especially in a hero play, and they are very proud of their performing art."
The individual kanji, from left to right, mean sing (歌), dance (舞), and skill (伎). Kabuki is therefore sometimes translated as "the art of singing and dancing".



