Hypoplastic Left Heart Syndrome

Hypoplastic left heart syndrome (HLHS) is a rare congenital heart defect in which the left side of the heart is severely underdeveloped. It may affect the left ventricle, aorta, aortic valve, or mitral valve.
Signs and symptoms
Closing of the ductus arteriosus in a heart that is severely underdeveloped on the left results in cyanosis and respiratory distress which can progress to cardiogenic shock and death. The first symptoms are cyanosis that does not respond to oxygen administration or poor feeding. Peripheral pulses may be weak and extremities cool to the touch.
HLHS often co-occurs with low birth weight and premature birth.
In neonates with a small atrial septal defect, termed "restrictive", there is inadequate mixing of oxygenated and deoxygenated blood. These neonates quickly decompensate and develop acidosis and cyanosis.
On EKG, right axis deviation and right ventricular hypertrophy are common, but not indicative of HLHS. Chest x-ray may show a large heart (cardiomegaly) or increased pulmonary vasculature. Neonates with HLHS do not typically have a heart murmur, but in some cases, a pulmonary flow murmur or tricuspid regurgitation murmur may be audible.
Co-occurring tricuspid regurgitation or right ventricular dysfunction can cause hepatomegaly to develop.
Causes

There is no known cause in the majority of HLHS cases. Some cases may have a genetic component, as HLHS has been shown to be heritable and associated with specific gene mutations.
Not all, but some, cases of aortic stenosis in a fetus can put stress on the left ventricle in utero, that can eventually lead to decreased perfusion and stop the growth of the left ventricle.
Environmental causes
In a retrospective analysis of over 1,300 newborns (born between 1996 and 2006) from 24 children’s hospitals in the United States, researchers at Cincinnati Children’s Hospital in Ohio found that babies with HLHS were more likely to be born in summer months, suggesting that seasonality and environmental factors may play a significant role in causation.
Genetics
Genetic loci associated with HLHS include GJA1 (connexin 43), HAND1, NKX2.5, 10q22, and 6q23. There is a slight risk of recurrence in future pregnancies, estimated to be 2-4%, which increases to 25% in families with two affected children. This is thought to be mediated by genetic mutations with incomplete penetrance.
HLHS is also associated with several genetic syndromes, including trisomy 13 (Patau syndrome), trisomy 18 (Edwards syndrome), partial trisomy 9, Turner's syndrome (XO), Jacobsen syndrome (11q deletion syndrome), Holt-Oram syndrome, and Smith-Lemli-Opitz syndrome.
Risk factors
Presence of a cystic hygroma increases the risk of HLHS in a fetus.
Pathophysiology
At birth, the ductus arteriosus is still open, and there is higher than normal resistance to blood flow in the lungs. This allows for adequate oxygenation via mixing between the atria and a normal appearance at birth. When the ductus begins to close and pulmonary vascular resistance decreases, blood flow through the ductus is restricted and flow to the lungs is increased.
In typical anatomy, the left side of the heart receives oxygen-rich blood from the lungs and pumps it to the rest of the body. In people with HLHS, the aorta and left ventricle are underdeveloped (beginning in utero), and the aortic and mitral valves are either too small to allow sufficient blood flow or are atretic (closed) altogether. As blood returns from the lungs to the left atrium, it cannot be pumped to the rest of the body by the left ventricle. The neonate is reliant on blood flowing through an atrial septal defect to mix oxygenated and deoxygenated blood, and on a patent ductus arteriosus to allow blood to reach the aorta and the systemic circulation via the right ventricle. This is what defines HLHS as a "single ventricle" defect.
Due to the underdevelopment of the left side of the heart in utero, the increased afterload causes hypertension of the left atrium, pulmonary edema, and therefore lung damage to the fetus before birth.
Diagnosis
Hypoplastic left heart syndrome can be diagnosed prenatally or after birth via echocardiography. Typical findings include a small left ventricle and aorta, abnormalities of the mitral and aortic valves, retrograde flow in the transverse arch of the aorta, and left-to-right flow between the atria. It is often recognized during the second trimester of pregnancy, between 18 and 24 weeks' gestation.
Management
Medical
Without life-prolonging interventions, HLHS is fatal, but with intervention, an infant may survive. A cardiothoracic surgeon may perform a series of operations or a full heart transplant. While surgical intervention has emerged as the standard of care in the United States, other national health systems, notably in France, approach diagnosis of HLHS in a more conservative manner, with an emphasis on termination of pregnancy or compassionate care after delivery.
Before surgery, the ductus must be kept open to allow blood-flow using medication containing prostaglandin. Air with less oxygen than normal is used for infants with hypoplastic left heart syndrome. These low oxygen levels increases the pulmonary vascular resistance (PVR) and thus improve blood flow to the rest of the body due to the greater pressure difference between the lungs and body. Achieving oxygen levels below atmosphere requires the use of inhaled nitrogen. Nitric oxide is a potent pulmonary vasodilator, and thus reduces PVR and improves venous return. Any factor that increases PVR will impede right sided flow.
Surgical
Surgical operations to assist with hypoplastic left heart are complex and need to be individualized for each patient. A cardiologist must assess all medical and surgical options on a case-by-case basis.
Currently, infants undergo either the staged reconstructive surgery (Norwood or Sano procedure within a few days of birth, Glenn or Hemi-Fontan procedure at 3 to 6 months of age, and the Fontan procedure at 1 1/2 to 5 years of age) or cardiac transplantation. Current expectations are that 70% of those with HLHS may reach adulthood. Many studies show that the higher the volume (number of surgeries performed) at a hospital, the lower the mortality (death) rate. Factors that increase an infant's risk include lower birth weight, additional congenital anomalies, a genetic syndrome or those with a highly restrictive atrial septum.) For patients without these additional risk factors, 5 year survival now approaches 80%. Studies show that about 75% of those children who survive surgery show developmental delays in one or more areas, such as motor, cognitive, or language impairments, with about a third of single-ventricle children without a genetic syndrome having significant impairments. Current research focuses on charting the connections between neurodevelopment injuries, surgical and intensive care procedures, and genetic susceptibility with the goal of modifying interventions that impair neurodevelopmental and psychosocial outcomes. An alternative to the traditional Norwood is the Hybrid procedure.
Some physicians offer compassionate care, instead of the surgeries, which results in the child's death, usually within 2 weeks of birth. Compassionate care is overseen by a physician, and may be carried out either in the hospital or at home. However, due to the vast improvement of surgical intervention, with many hospitals achieving over 90% survival, there is debate on whether or not compassionate care should still be offered to families. A study in 2003 concluded that a selection of physicians who are experts in the care of children with HLHS were evenly split when asked what they would do if their own children were born with HLHS, with 1/3 stating that they would choose surgery, 1/3 stating that they would choose palliative (compassionate) treatment without surgery, and 1/3 stating that they are uncertain which choice they would make.
The three-stage procedure is a palliative procedure (not a cure), as the child's circulation is made to work with only two of the heart's four chambers.
Norwood procedure
The first step is the Norwood procedure. In this procedure, the right ventricle is used to pump blood into the systemic circulation. Since the right ventricle is no longer directly pumping blood to the lungs, a shunt is required in order to pass deoxygenated blood through the lungs. Either the subclavian artery can be connected to the pulmonary circulation (Blalock-Taussig shunt), or a shunt is made directly from the right ventricle to the pulmonary circulation (Sano shunt). The narrow aorta is enlarged using a patch to improve blood flow to the body.
During this time the baby may be medically fragile and have feeding problems because the heart is working very hard. There is a considerable degree of venous mixing in the right ventricle, leading to lower oxygenation saturation. In addition, both the Blalock-Taussig and the Sano shunts expose the lungs to systemic arterial pressures, leading to long-term pulmonary hypertension and eventually heart failure.
Hybrid procedure
The Hybrid procedure may be used in place of the Norwood. The Hybrid procedure does not necessitate the use of heart-lung bypass or performing a sternotomy. Instead of a six-hour surgery, the Hybrid typically takes one to two hours. In this procedure, a stent is placed in the ductus arteriosus to maintain its patency, and bands are placed over both the left and right pulmonary artery branches to limit pressure and over-circulation to the lungs. Outcomes with the Hybrid approach are comparable to those with the Norwood.
Glenn procedure
The second stage—the bidirectional Glenn or Hemi-Fontan (see also Kawashima procedure)—relieves some of the problems introduced by Stage I palliation. In this operation, the superior vena cava is ligated from the heart and connected to the pulmonary circulation. At this time, the Blalock-Taussig or Sano shunt is taken down. The lungs are no longer exposed to systemic arterial pressures, but much lower venous pressures. Although venous blood from the upper half of the body is no longer mixing with oxygenated blood in the right ventricle, there is still venous mixing from the lower half of the body, leading to some degree of oxygen desaturation.
Fontan procedure
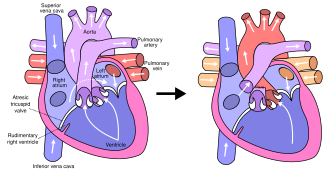
The final procedure, the Fontan procedure, completes the repair of the hypoplastic left heart. Although there are several variations, the functional effect is to redirect venous blood from the lower body (through the inferior vena cava) away from the right atrium to the pulmonary artery. This should eliminate any mixing of oxygenated and deoxygenated blood in the right ventricle. The right ventricle performs the traditional job of the left, supplying the body with oxygenated blood, while the passive systemic venous pressure performs the traditional job of the right, passing deoxygenated blood to the lungs.
Fetal surgery
Interventions performed during fetal development are under investigation. In fetuses with hypoplastic left ventricles and an intact interatrial septum, percutaneous atrial septostomy has been attempted.
Prognosis
95% of untreated infants with HLHS die in the first weeks of life.
Early survival has improved since the introduction of the Norwood procedure. Since there are no long-term studies of HLHS adults, statistics are usually derived from post-Fontan patients; it is estimated that 70% of HLHS patients may reach adulthood.
Prognosis is dependent upon the health of the child, as there is an increased demand on respiratory and heart rate in infants during common childhood illnesses. This fragile population has little cardiac reserve to accommodate these demands and provide hemodynamic stability during illnesses.
Children with HLHS and other comparable single-ventricle conditions, as a group, have poorer neurodevelopmental outcomes than their healthy peers. Deficits in language, executive functioning, and higher rates of anxiety and depression disorders have been demonstrated. Some of these outcomes may be a consequence of genetic factors associated with HLHS, and others may be modifiable through changes to procedures and to the healthcare environment. There is an emerging clinical consensus around the importance of continuous neurodevelopmental surveillance from the earliest years into adulthood.
As is true for patients with other types of heart defects involving malformed valves, HLHS patients run a high risk of endocarditis, and must be monitored by a cardiologist for the rest of their lives to check on their heart function.
Heart transplantation may be indicated, typically after Fontan completion. One multi-center study (of patients undergoing the Fontan from 1993-2001) reported a 76% 1-year survival rate in patients who survived to transplant.
Epidemiology
HLHS occurs in an estimated 1 out of 4,300 live births in the United States, or an estimated total of 960 live births per year in that country. Overall, it is estimated to make up 2-3% of all cases of congenital heart disease, and is the most common single-ventricle defect. It is thought to be more common in male infants, 1.5 times as common as in female infants.